Scroll left to select an efficacy measure
Progression-Free Survival in mTNBC (HR-/HER2-)
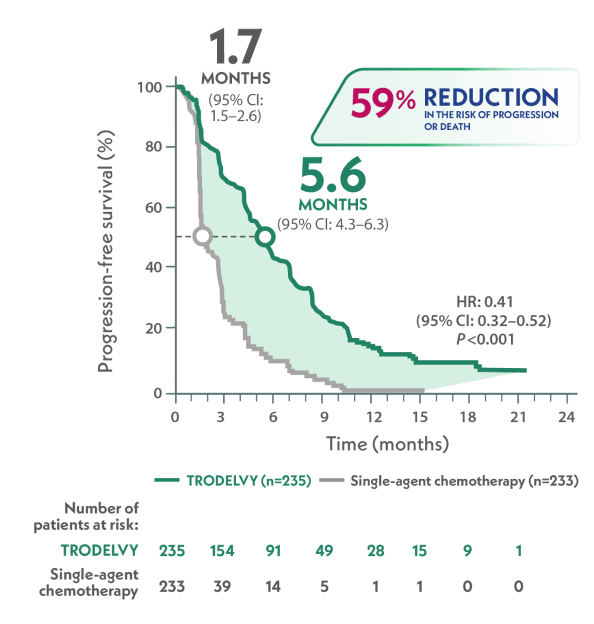
Statistically significant and clinically meaningful mPFS benefit1
Median Progression-free Survival
Proven mPFS benefit in a landmark Phase 3 clinical trial with over 500 patients1,2
TRODELVY was studied in ASCENT, a Phase 3, randomized, active-controlled, open-label study (N=529). The efficacy analysis included progression-free survival (PFS) in brain-met–negative patients (primary endpoint) by BICR based on RECIST 1.1 criteria, with PFS for the full population (all patients with and without brain metastases) and overall survival (OS) as secondary endpoints.1,2 88% of patients in the full population were brain-met–negative.1,2
A phase 3, randomized, active-controlled, open-label study
Scroll left to review

Single-agent chemotherapy was determined by the investigator before randomization from one of the following choices: eribulin (n=139), capecitabine (n=33), gemcitabine (n=38), or vinorelbine (n=52).1
Patients with brain metastases were allowed to enroll up to a predefined maximum of 15% of patients in the ASCENT study; MRI was required prior to enrollment for patients with known or suspected brain metastases. Patients with known Gilbert’s disease or bone-only disease were excluded.1
aAll patients received previous taxane treatment in either the adjuvant, neoadjuvant, or advanced stage unless there was a contraindication or intolerance to taxanes during or at the end of the first taxane cycle.1
The population of ASCENT has characteristics that may resemble those of your patients1
DEMOGRAPHICS1 |
|
|---|---|
| Median age: | 54 years (range: 27−82 years); 81% <65 years |
| Sex: | 99.6% female |
| Race/ethnicity: | 79% White; 12% Black/African American |
DISEASE CHARACTERISTICS1 |
|
|---|---|
| Hepatic metastases (visceral disease): | 42% |
| Brain metastases: | 12% |
| BRCA1/BRCA2 positive: | 9% |
| ECOG performance status: | 0 (43%); 1 (57%) |
TREATMENT HISTORY1 |
|
|---|---|
| Prior PD-1/PD-L1 therapy: | 29% |
| 1 prior line of systemic therapy in metastatic settingb: | 13% in TRODELVY group |
bIn addition to having disease recurrence or progression within 12 months of neoadjuvant/adjuvant systemic therapy.1
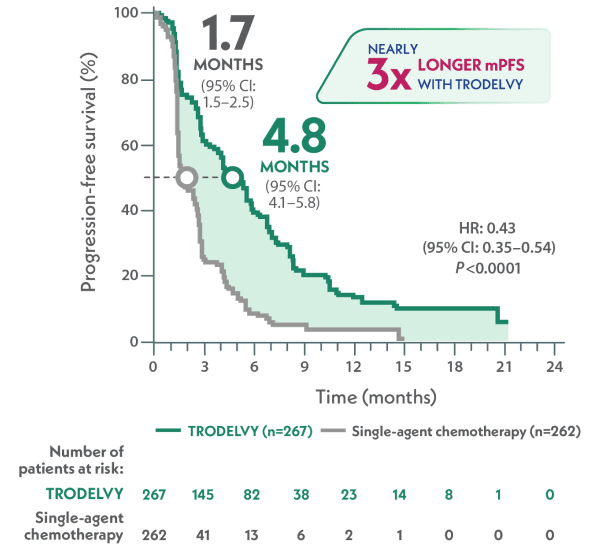
More than 3x longer median PFS with TRODELVY compared to single-agent chemotherapy in 2L+ mTNBC2
Primary Endpoint: mPFS in the brain-met–negative population by BICR per RECIST 1.1 criteria2,c
>3x LONGER
VS
HR: 0.41 (95% CI: 0.32–0.52); P<0.001
TRODELVY was studied in patients across IHC status (HER2-low and IHC 0)3
cPFS was defined as the time from the date of randomization to the date of the first radiological disease progression or death due to any cause, whichever came first.1
Nearly 3x longer median PFS with TRODELVY compared to single-agent chemotherapy1,2
mPFS in the full population by BICR per RECIST 1.1 criteria1,2,d
~3x LONGER
VS
HR: 0.43 (95% CI: 0.35–0.54); P<0.001
dPFS is defined as the time from the date of randomization to the date of the first radiological disease progression or death due to any cause, whichever came first.1
TRODELVY is the only Trop-2–directed ADC with statistically significant PFS compared to single-agent chemotherapy in 2L+ mTNBC1
Discuss the data further with your
Gilead representative
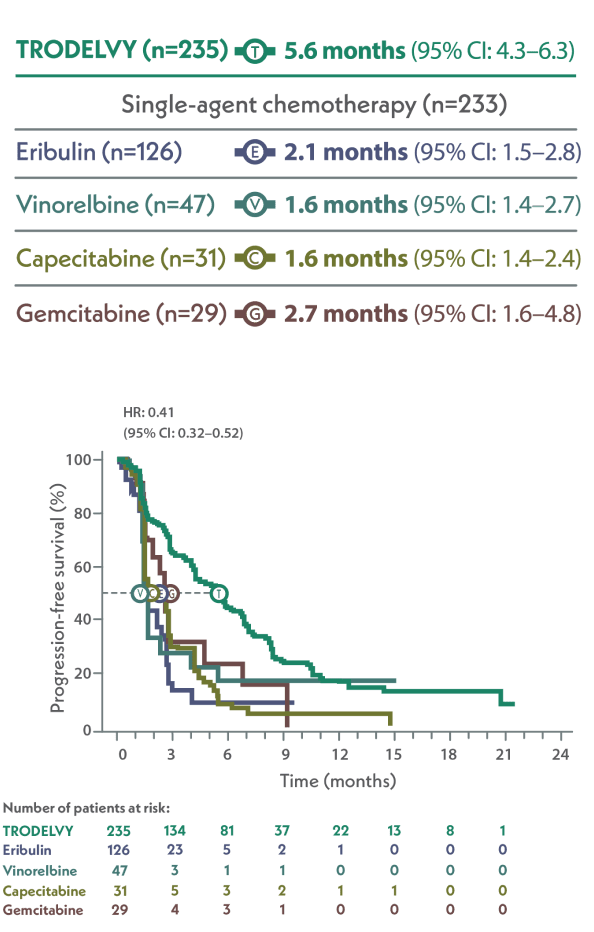
In a subgroup analysis of ASCENT:
PFS across subgroups in the primary analysis population
Limitation: These results are from a subgroup analysis of the Phase 3 ASCENT study. This secondary endpoint was not powered for statistical analysis and should be considered descriptive only. Therefore, the results require cautious interpretation and could represent chance findings.
mPFS by BICR based on RECIST 1.1 criteria and hazard ratio for disease progression or death (brain-met–negative population)2
Scroll left to review
88% of patients in the ASCENT study were brain-met–negative, and PFS results of this subanalysis were consistent with the ASCENT primary findings.1,2
Reproduced with permission from Bardia et al. N Engl J Med. 2021; copyright Massachusetts Medical Society.
In a subgroup analysis of ASCENT:
mPFS of TRODELVY vs single-agent chemotherapy from ASCENT4
Limitation: These results are from a post hoc subgroup analysis of the Phase 3 ASCENT study. The single-agent chemotherapy arms were not powered for statistical analysis or designed to compare against individual agents, and findings should be considered descriptive only. Therefore, the results require cautious interpretation and could represent chance findings.4
mPFS in the brain-met–negative population by BICR per RECIST 1.1 criteriae

88% of patients in the ASCENT study were brain-met–negative, and PFS results of this subanalysis were consistent with the ASCENT primary findings.1,2
ePFS was defined as the time from the date of randomization to the date of the first radiological disease progression or death due to any cause, whichever came first.1
Select safety findings4
- Key Grade ≥3 treatment-related adverse events (TRAEs) with TRODELVY vs eribulin included neutropenia (51% vs 31%), leukopenia (10% vs 5%), diarrhea (10% vs 0%), anemia (8% vs 2%), febrile neutropenia (6% vs 2%), fatigue (3% vs 5%), nausea (3% vs 1%), and vomiting (1% vs 1%)
- Key Grade ≥3 TRAEs with TRODELVY vs vinorelbine, capecitabine, and gemcitabine combined included neutropenia (51% vs 36%), leukopenia (10% vs 6%), diarrhea (10% vs 1%), anemia (8% vs 8%), febrile neutropenia (6% vs 2%), fatigue (3% vs 6%), nausea (3% vs 0%), and vomiting (1% vs 0%)
- Discontinuation rates due to treatment-emergent adverse events for TRODELVY, eribulin, vinorelbine, capecitabine, and gemcitabine were 5%, 2%, 10%, 7%, and 9%, respectively
- One treatment-related death was reported for the single-agent chemotherapy arm (eribulin; neutropenic sepsis) and none with TRODELVY
ASCENT TRIAL VIDEO OVERVIEW
Summary of ASCENT Efficacy and Safety
With Dr. Yuan Yuan, MD, PhD
<TEXT ON-SCREEN>
TRODELVY®
sacituzumab govitecan-hziy
180 mg for injection
Continue watching for Important Safety Information at the end of this video.
Please see link provided for full Prescribing Information, including BOXED WARNING.
TRODELVY® in mTNBC
mTNBC=metastatic triple-negative breast cancer.
<END OF TEXT ONSCREEN>
<VOICEOVER AND TEXT ON-SCREEN>
INDICATION: TRODELVY® (sacituzumab govitecan-hziy) is a Trop-2-directed antibody and topoisomerase inhibitor conjugate indicated for the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (mTNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease.
IMPORTANT SAFETY INFORMATION
BOXED WARNING: NEUTROPENIA AND DIARRHEA
- Severe or life-threatening neutropenia may occur. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Consider G-CSF for secondary prophylaxis. Initiate anti-infective treatment in patient with febrile neutropenia without delay.
- Severe diarrhea may occur. Monitor patients with diarrhea and give fluid and electrolytes as needed. At the onset of diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤Grade 1 and reduce subsequent doses.
Continue watching for Important Safety Information at the end of this video.
<END OF VOICEOVER AND TEXT ONSCREEN>
DR. YUAN: Metastatic triple-negative breast cancer is an aggressive disease with poor overall survival. When compared with other types of breast cancer following metastatic diagnosis, the survival probability of triple-negative breast cancer is somewhat lower.
<TEXT ON-SCREEN>
Dr. Yuan Yuan
Cedars-Sinai Cancer Center
<END OF TEXT ONSCREEN>
DR. YUAN: For patients with metastatic triple-negative breast cancer, the five-year relative survival rate is only 12%, the worst of all breast cancer subtypes.
Given all of this, there is a significant need for effective treatments that could prolong survival in patients with metastatic triple-negative breast cancer.
<TEXT ON-SCREEN>
An option you can offer your patient
<END OF TEXT ONSCREEN>
DR. YUAN: Considering how triple-negative breast cancer, especially in the metastatic setting, how aggressive the disease is, I think the primary goal is to be able to offer them an option that extends the overall survival and delay the progression.
<TEXT ON-SCREEN>
As early as second line1
<END OF TEXT ONSCREEN>
DR. YUAN: TRODELVY, also known as sacituzumab govitecan-hziy, is an option you can offer your patient as early as second-line treatment for metastatic triple-negative breast cancer following the use of two or more prior systemic therapies, at least one of them for metastatic disease.
DR. YUAN: The efficacy and safety of TRODELVY were studied in ASCENT, a landmark Phase 3 trial assessing survival in more than 500 patients with pretreated metastatic triple-negative breast cancer. Patients in ASCENT had relapsed after at least two prior chemotherapies, at least one of them for metastatic disease.
<TEXT ON-SCREEN>
In this randomized, active-controlled, open-label study, TRODELVY was evaluated versus single-agent chemotherapy1,2
Patient population (N=529)*
Patients with unresectable locally advanced or mTNBC who had relapsed after at least 2 prior chemotherapies, at least one of them for metastatic disease (one of which could be in the neoadjuvant or adjuvant setting provided progression occurred within a 12-month period)
1:1 RANDOMIZATION
TRODELVY 10 mg/kg IV on Days 1 and 8 of a 21-day cycle (n=267)
Single-agent chemotherapy (n=262)
ENDPOINTS
Primary
PFS in brain-met−negative population by BICR based on RECIST 1.1 criteria
Select secondary
PFS (investigator assessment)
OS
ORR
Safety
*All patients received previous taxane treatment in either the adjuvant, neoadjuvant, or advanced stage unless there was a contraindication or intolerance to taxanes during or at the end of the first taxane cycle.
BICR=blinded independent central review; IV=intravenous; ORR=objective response rate; OS=overall survival; PFS=progression-free survival; RECIST=Response Evaluation Criteria in Solid Tumors.
<END OF TEXT ONSCREEN>
DR. YUAN: The primary endpoint was progression-free survival in the brain metastasis–free population, as assessed by blinded independent central review per RECIST 1.1 criteria. Secondary endpoints include progression-free survival in the full population, overall survival in both the brain metastasis–negative and full populations, and objective response rate.
Given the need for therapies that could prolong survival, consider the outcomes data from ASCENT, which I will talk about in a moment, as you plan the next step in your patient’s treatment plans.
<TEXT ON-SCREEN>
Kaplan-Meier estimates of mPFS by BICR based on RECIST 1.1 criteria (full population)1*
mTNBC
mPFS1
4.8 months with TRODELVY
(95% CI: 4.1–5.8)(n=267)
Vs
1.7 months with chemotherapy
(95% CI: 1.5–2.5)(n=262)
57% reduction in the risk of disease progression or death
HR: 0.43 (95% CI: 0.35–0.54); P<0.0001
Primary endpoint: In the primary analysis (brain-met–negative) population, TRODELVY demonstrated statistically significant mPFS results versus single-agent chemotherapy2
mPFS was 5.6 months with TRODELVY (95% CI: 4.3–6.3)(n=235) versus 1.7 months with single-agent chemotherapy (95% CI: 1.5–2.6)(n=233); HR: 0.41 (95% CI: 0.32–0.52); P<0.001
Exploratory findings in previously treated, stable brain-met–positive patients1
mPFS was 2.8 months with TRODELVY (95% CI: 1.5–3.9) versus 1.6 months with single-agent chemotherapy (95% CI: 1.3–2.9); HR: 0.65 (95% CI: 0.35–1.22)
*PFS is defined as the time from the dates of randomization to the date of the first radiological disease progression or death due to any cause, whichever comes first.1
CI=confidence interval; HR=hazard ratio; mPFS=median progression-free survival.
<END OF TEXT ONSCREEN>
DR. YUAN: In ASCENT, TRODELVY demonstrated median progression-free survival that was nearly three times longer than that with single-agent chemotherapy. Patients taking TRODELVY in the full population had a median progression-free survival of 4.8 months versus 1.7 months with single-agent chemotherapy. This represents a 57% reduction in the risk of disease progression or death.
<TEXT ON-SCREEN>
Kaplan-Meier estimates of mPFS by BICR based on RECIST 1.1 criteria (full population)1*
mTNBC
mPFS1
4.8 months with TRODELVY
(95% CI: 4.1–5.8)(n=267)
Vs
1.7 months with chemotherapy
(95% CI: 1.5–2.5)(n=262)
57% reduction in the risk of disease progression or death
HR: 0.43 (95% CI: 0.35–0.54); P<0.0001
Primary endpoint: In the primary analysis (brain-met–negative) population, TRODELVY demonstrated statistically significant mPFS results versus single-agent chemotherapy2
mPFS was 5.6 months with TRODELVY (95% CI: 4.3–6.3)(n=235) versus 1.7 months with single-agent chemotherapy (95% CI: 1.5–2.6)(n=233); HR: 0.41 (95% CI: 0.32–0.52); P<0.001
Exploratory findings in previously treated, stable brain-met–positive patients1
mPFS was 2.8 months with TRODELVY (95% CI: 1.5–3.9) versus 1.6 months with single-agent chemotherapy (95% CI: 1.3–2.9); HR: 0.65 (95% CI: 0.35–1.22)
*PFS is defined as the time from the dates of randomization to the date of the first radiological disease progression or death due to any cause, whichever comes first.1
Kaplan-Meier estimates of median OS (full population)1
mTNBC
mOS1
11.8 months with TRODELVY
(95% CI: 10.5–13.8)(n=267)
Vs
6.9 months with chemotherapy
(95% CI: 5.9–7.6)(n=262)
49% reduction in the risk of death
HR: 0.51 (95% CI: 0.41–0.62); P<0.0001
In the primary analysis (brain-met–negative) population, TRODELVY demonstrated statistically significant improvement in mOS results versus single-agent chemotherapy2
mOS was 12.1 months with TRODELVY (95% CI: 10.7–14.0) (n=235) versus 6.7 months with single-agent chemotherapy (95% CI: 0.38–0.59); P<0.001
Exploratory findings in previously treated, stable brain-met–positive patients1
mOS was 6.8 months with TRODELVY (95% CI: 4.7–14.1) versus 7.4 months with single-agent chemotherapy (95% CI: 4.7–11.1); HR: 0.87 (95% CI: 0.47–1.63)
mOS=median overall survival.
<END OF TEXT ON-SCREEN>
DR. YUAN: In the primary analysis of brain metastasis–negative population, TRODELVY demonstrated statistically significant progression-free survival with a median of 5.6 months compared to 1.7 months with single-agent chemotherapy.
In terms of overall survival, TRODELVY had a median overall survival of 11.8 months in the full population of ASCENT, vs 6.9 months with single-agent chemotherapy, representing a 49% reduction in the risk of death.
<TEXT ON-SCREEN>
TRODELVY is the only ADC approved to provide a statistically significant overall survival improvement in mTNBC1
ADC=antibody-drug conjugate.
<END OF TEXT ON-SCREEN>
DR. YUAN: TRODELVY is the only ADC to provide a statistically significant overall survival improvement in metastatic triple-negative breast cancer. TRODELVY could be your next opportunity to delay disease progression and extend survival in metastatic triple-negative breast cancer.
DR. YUAN: In the ASCENT trial, adverse reactions that led to discontinuation of TRODELVY occurred in 5% of patients. Adverse reactions leading to permanent discontinuation in at least 1% of patients who received TRODELVY were pneumonia, 1%; fatigue, 1%.
Serious adverse reactions occurred in 27% of patients receiving TRODELVY.
<TEXT ON-SCREEN>
Adverse reactions in ≥10% of patients with mTNBC in the ASCENT trial1
Adverse reactions that led to discontinuation of TRODELVY occurred in 5% of patients1
Adverse reactions leading to permanent discontinuation in ≥1% of patients who received TRODELVY were pneumonia (1%) and fatigue (1%)1
Serious adverse reactions occurred in 27% of patients receiving TRODELVY
Serious adverse reactions in >1% of patients receiving TRODELVY included neutropenia (7%), diarrhea (4%), and pneumonia (3%)
The most common (≥25%) adverse reactions, including lab abnormalities, were decreased hemoglobin (94%), decreased lymphocyte count (88%), decreased leukocyte count (86%), decreased neutrophil count (78%), fatigue (65%), diarrhea (59%), nausea (57%), increased glucose (49%), alopecia (47%), constipation (37%), decreased calcium (36%), vomiting (33%), decreased magnesium (33%), decreased potassium (33%), increased albumin (32%), abdominal pain (30%), decreased appetite (28%), increased aspartate aminotransferase (27%), increased alanine aminotransferase (26%), increased alkaline phosphatase (26%), and decreased phosphate (26%)
For information on select laboratory abnormalities, please refer to Table 3 of the full Prescribing Information.
Graded per NCI CTCAE v5.0.1
aSingle-agent chemotherapy included one of the following single agents: eribulin (n=139), capcitabine (n=33), gemcitabine (n=38), or vinorelbine (except if patients had ≥Grade 2 neuropathy, n=52).1
bIncluding stomatitis, glossitis, mouth ulceration, and mucosal inflammation.1
cIncluding fatigue and asthenia.1
NCI CTCAE=National Cancer Institution Common Terminology Criteria for Adverse Events.
<END OF TEXT ON-SCREEN>
DR. YUAN: Serious adverse reactions in over 1% of patients receiving TRODELVY included neutropenia, 7%; diarrhea, 4%; and pneumonia, 3%.
The most common adverse reactions in ASCENT were fatigue, 65%; neutropenia, 64%; diarrhea, 59%; nausea, 57%; alopecia, 47%; anemia, 40%; constipation, 37%; vomiting, 33%; abdominal pain, 30%; and decreased appetite, 28%.
<TEXT ON-SCREEN>
TRODELVY offers you the opportunity to elevate survival2
Dr. Yuan Yuan
Cedars-Sinai Cancer Center
<END OF TEXT ON-SCREEN>
DR. YUAN: As we have seen, TRODELVY offers you the opportunity to elevate survival. This is an important development in the treatment landscape.
<TEXT ON-SCREEN>
You have the potential to extend survival
Choose TRODELVY in mTNBC
<END OF TEXT ON-SCREEN>
<VOICEOVER AND TEXT ON-SCREEN>
INDICATION: TRODELVY® (sacituzumab govitecan-hziy) is a Trop-2-directed antibody and topoisomerase inhibitor conjugate indicated for the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (mTNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease.
IMPORTANT SAFETY INFORMATION
BOXED WARNING: NEUTROPENIA AND DIARRHEA
- TRODELVY can cause severe, life-threatening, or fatal neutropenia. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Primary prophylaxis with G-CSF is recommended for all patients at increased risk of febrile neutropenia. Initiate anti-infective treatment in patients with febrile neutropenia without delay.
- TRODELVY can cause severe diarrhea. Monitor patients with diarrhea and give fluid and electrolytes as needed. At the onset of diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤Grade 1 and reduce subsequent doses.
CONTRAINDICATIONS
- Severe hypersensitivity reaction to TRODELVY.
WARNINGS AND PRECAUTIONS
Neutropenia: Severe, life-threatening, or fatal neutropenia can occur as early as the first cycle of treatment and may require dose modification. Neutropenia occurred in 64% of patients treated with TRODELVY. Grade 3-4 neutropenia occurred in 49% of patients. Febrile neutropenia occurred in 6%. Neutropenic colitis occurred in 1.4%. Primary prophylaxis with G-CSF is recommended starting in the first cycle of treatment in all patients at increased risk of febrile neutropenia, including older patients, patients with previous neutropenia, poor performance status, organ dysfunction, or multiple comorbidities. Monitor absolute neutrophil count (ANC) during treatment. Withhold TRODELVY for ANC below 1500/mm3 on Day 1 of any cycle or below 1000/mm3 on Day 8 of any cycle. Withhold TRODELVY for neutropenic fever. Treat neutropenia with G-CSF and administer prophylaxis in subsequent cycles as clinically indicated or indicated in Table 2 of USPI.
Diarrhea: Diarrhea occurred in 64% of all patients treated with TRODELVY. Grade 3-4 diarrhea occurred in 11% of patients. One patient had intestinal perforation following diarrhea. Diarrhea that led to dehydration and subsequent acute kidney injury occurred in 0.7% of all patients. Withhold TRODELVY for Grade 3-4 diarrhea and resume when resolved to ≤Grade 1. At onset, evaluate for infectious causes and if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (e.g., fluid and electrolyte substitution) may also be employed as clinically indicated. Patients who exhibit an excessive cholinergic response to treatment can receive appropriate premedication (e.g., atropine) for subsequent treatments.
Hypersensitivity and Infusion-Related Reactions: TRODELVY can cause serious hypersensitivity reactions including life-threatening anaphylactic reactions. Severe signs and symptoms included cardiac arrest, hypotension, wheezing, angioedema, swelling, pneumonitis, and skin reactions. Hypersensitivity reactions within 24 hours of dosing occurred in 35% of patients. Grade 3-4 hypersensitivity occurred in 2% of patients. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.2%. The incidence of anaphylactic reactions was 0.2%. Pre-infusion medication is recommended. Have medications and emergency equipment to treat such reactions available for immediate use. Observe patients closely for hypersensitivity and infusion-related reactions during each infusion and for at least 30 minutes after completion of each infusion. Permanently discontinue TRODELVY for Grade 4 infusion-related reactions.
Nausea and Vomiting: TRODELVY is emetogenic and can cause severe nausea and vomiting. Nausea occurred in 64% of all patients treated with TRODELVY and Grade 3-4 nausea occurred in 3% of these patients. Vomiting occurred in 35% of patients and Grade 3-4 vomiting occurred in 2% of these patients. Premedicate with a two or three drug combination regimen (e.g., dexamethasone with either a 5-HT3 receptor antagonist or an NK1 receptor antagonist as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting (CINV). Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting and resume with additional supportive measures when resolved to Grade ≤1. Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity: Patients homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia and may be at increased risk for other adverse reactions with TRODELVY. The incidence of Grade 3-4 neutropenia was 58% in patients homozygous for the UGT1A1*28 allele, 49% in patients heterozygous for the UGT1A1*28 allele, and 43% in patients homozygous for the wild-type allele. The incidence of Grade 3-4 anemia was 21% in patients homozygous for the UGT1A1*28 allele, 10% in patients heterozygous for the UGT1A1*28 allele, and 9% in patients homozygous for the wild-type allele. Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on clinical assessment of the onset, duration and severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 function.
Embryo-Fetal Toxicity: Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
ADVERSE REACTIONS
In the pooled safety population, the most common (≥25%) adverse reactions including laboratory abnormalities were decreased leukocyte count (84%), decreased neutrophil count (75%), decreased hemoglobin (69%), diarrhea (64%), nausea (64%), decreased lymphocyte count (63%), fatigue (51%), alopecia (45%), constipation (37%), increased glucose (37%), decreased albumin (35%), vomiting (35%), decreased appetite (30%), decreased creatinine clearance (28%), increased alkaline phosphatase (28%), decreased magnesium (27%), decreased potassium (26%), and decreased sodium (26%).
In the ASCENT study (locally advanced or metastatic triple-negative breast cancer), the most common adverse reactions (incidence ≥25%) were fatigue, diarrhea, nausea, alopecia, constipation, vomiting, abdominal pain, and decreased appetite. The most frequent serious adverse reactions (SAR) (>1%) were neutropenia (7%), diarrhea (4%), and pneumonia (3%). SAR were reported in 27% of patients, and 5% discontinued therapy due to adverse reactions. The most common Grade 3-4 lab abnormalities (incidence ≥25%) in the ASCENT study were reduced neutrophils, leukocytes, and lymphocytes.
DRUG INTERACTIONS
UGT1A1 Inhibitors: Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38. Avoid administering UGT1A1 inhibitors with TRODELVY.
UGT1A1 Inducers: Exposure to SN-38 may be reduced in patients concomitantly receiving UGT1A1 enzyme inducers. Avoid administering UGT1A1 inducers with TRODELVY.
Please see full Prescribing Information, including BOXED WARNING.
References: 1. Prescribing Information. Gilead Sciences, Inc.; March 2025. 2. Bardia A, Hurvitz SA, Tolaney SM, et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N Engl J Med. 2021;384(16):1529-1541. 3. Bardia A, Tolaney SM, Loirat D, et al. Sacituzumab govitecan versus treatment of physician's choice in patients with previously treated metastatic triple-negative breast cancer: final data from the phase 3 ASCENT study. Poster presented at: American Society of Clinical Oncology Annual Meeting; June 3-7, 2022; Chicago, IL. Poster 1071.
<END OF VOICEOVER AND TEXT ON-SCREEN>
<TEXT ON-SCREEN>
TRODELVY®
sacituzumab govitecan-hziy
180 mg for injection
GILEAD Oncology
TRODELVY, the TRODELVY logo, GILEAD, and the GILEAD logo are trademarks of Gilead Sciences, Inc., or its related companies. All other marks are the property of their respective owners.
© 2025 Gilead Sciences, Inc. All rights reserved. US-TROP-2029 06/25
<END OF TEXT ON-SCREEN>
2L=second-line; ADC=antibody-drug conjugate; BICR=blinded independent central review; brain-met=brain metastases; CI=confidence interval; ECOG= Eastern Cooperative Oncology Group; HER2=human epidermal growth factor receptor 2; HR=hazard ratio; IHC=immunohistochemistry; mPFS=median progression-free survival; mTNBC=metastatic triple-negative breast cancer; NE=not evaluable; PD-1=programmed death 1 protein; PD-L1=programmed death ligand 1; PFS=progression-free survival; RECIST=Response Evaluation Criteria in Solid Tumors; TNBC=triple-negative breast cancer; yr=year.
References: 1. TRODELVY. Prescribing Information. Gilead Sciences, Inc.; March 2025. 2. Bardia A, Hurvitz SA, Tolaney SM, et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N Engl J Med. 2021;384(16):1529-1541. 3. Immunomedics, Inc. An international, multi-center, open-label, randomized, phase III trial of sacituzumab govitecan versus treatment of physician choice in patients with metastatic triple-negative breast cancer who received at least two prior treatments. Published November 18, 2015. Updated June 22, 2017. Accessed April 30, 2025. https://www.nejm.org/doi/suppl/l0.1056/NEJMoa2028485/suppl_file/nejmoa2028485_protocol.pdf 4. O’Shaughnessy J, Punie K, Oliveira M, et al. Assessment of sacituzumab govitecan vs treatment of physician’s choice cohort by agent in the phase 3 ASCENT study of patients with metastatic triple-negative breast cancer. Poster presented at: Virtual American Society of Clinical Oncology (ASCO) Annual Meeting; June 4-8, 2021. Poster 1077. https://meetings.asco.org/meetings/2021-asco-annual-meeting/273/program-guide/scheduled-sessions
Continue exploring mTNBC efficacy
Select another topic
Scroll left to select an efficacy measure

TRODELVY® (sacituzumab govitecan-hziy) is a Trop-2-directed antibody and topoisomerase inhibitor conjugate indicated for the treatment of adult patients with:
- Unresectable locally advanced or metastatic triple-negative breast cancer (mTNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease.
- Unresectable locally advanced or metastatic hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting.
Important Safety Information
Tap for Important Safety Information, including BOXED WARNING: Neutropenia and Diarrhea.
Boxed Warning: neutropenia and diarrhea
- TRODELVY can cause severe, life-threatening, or fatal neutropenia. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Primary prophylaxis with G-CSF is recommended for all patients at increased risk of febrile neutropenia. Initiate anti-infective treatment in patients with febrile neutropenia without delay.
- TRODELVY can cause severe diarrhea. Monitor patients with diarrhea and give fluid and electrolytes as needed. At the onset of diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤Grade 1 and reduce subsequent doses.
Contraindications
- Severe hypersensitivity reaction to TRODELVY.
Warnings and precautions
Neutropenia: Severe, life-threatening, or fatal neutropenia can occur as early as the first cycle of treatment and may require dose modification. Neutropenia occurred in 64% of patients treated with TRODELVY. Grade 3-4 neutropenia occurred in 49% of patients. Febrile neutropenia occurred in 6%. Neutropenic colitis occurred in 1.4%. Primary prophylaxis with G-CSF is recommended starting in the first cycle of treatment in all patients at increased risk of febrile neutropenia, including older patients, patients with previous neutropenia, poor performance status, organ dysfunction, or multiple comorbidities. Monitor absolute neutrophil count (ANC) during treatment. Withhold TRODELVY for ANC below 1500/mm3 on Day 1 of any cycle or below 1000/mm3 on Day 8 of any cycle. Withhold TRODELVY for neutropenic fever. Treat neutropenia with G-CSF and administer prophylaxis in subsequent cycles as clinically indicated or indicated in Table 2 of USPI.
Diarrhea: Diarrhea occurred in 64% of all patients treated with TRODELVY. Grade 3-4 diarrhea occurred in 11% of patients. One patient had intestinal perforation following diarrhea. Diarrhea that led to dehydration and subsequent acute kidney injury occurred in 0.7% of all patients. Withhold TRODELVY for Grade 3-4 diarrhea and resume when resolved to ≤Grade 1. At onset, evaluate for infectious causes and if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (e.g., fluid and electrolyte substitution) may also be employed as clinically indicated. Patients who exhibit an excessive cholinergic response to treatment can receive appropriate premedication (e.g., atropine) for subsequent treatments.
Hypersensitivity and Infusion-Related Reactions: TRODELVY can cause serious hypersensitivity reactions including life-threatening anaphylactic reactions. Severe signs and symptoms included cardiac arrest, hypotension, wheezing, angioedema, swelling, pneumonitis, and skin reactions. Hypersensitivity reactions within 24 hours of dosing occurred in 35% of patients. Grade 3-4 hypersensitivity occurred in 2% of patients. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.2%. The incidence of anaphylactic reactions was 0.2%. Pre-infusion medication is recommended. Have medications and emergency equipment to treat such reactions available for immediate use. Observe patients closely for hypersensitivity and infusion-related reactions during each infusion and for at least 30 minutes after completion of each infusion. Permanently discontinue TRODELVY for Grade 4 infusion-related reactions.
Nausea and Vomiting: TRODELVY is emetogenic and can cause severe nausea and vomiting. Nausea occurred in 64% of all patients treated with TRODELVY and Grade 3-4 nausea occurred in 3% of these patients. Vomiting occurred in 35% of patients and Grade 3-4 vomiting occurred in 2% of these patients. Premedicate with a two or three drug combination regimen (e.g., dexamethasone with either a 5-HT3 receptor antagonist or an NK1 receptor antagonist as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting (CINV). Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting and resume with additional supportive measures when resolved to Grade ≤1. Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity: Patients homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia and may be at increased risk for other adverse reactions with TRODELVY. The incidence of Grade 3-4 neutropenia was 58% in patients homozygous for the UGT1A1*28, 49% in patients heterozygous for the UGT1A1*28 allele, and 43% in patients homozygous for the wild-type allele. The incidence of Grade 3-4 anemia was 21% in patients homozygous for the UGT1A1*28 allele, 10% in patients heterozygous for the UGT1A1*28 allele, and 9% in patients homozygous for the wild-type allele. Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on clinical assessment of the onset, duration and severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 function.
Embryo-Fetal Toxicity: Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
Adverse Reactions
In the pooled safety population, the most common (≥25%) adverse reactions including laboratory abnormalities were decreased leukocyte count (84%), decreased neutrophil count (75%), decreased hemoglobin (69%), diarrhea (64%), nausea (64%), decreased lymphocyte count (63%), fatigue (51%), alopecia (45%), constipation (37%), increased glucose (37%), decreased albumin (35%), vomiting (35%), decreased appetite (30%), decreased creatinine clearance (28%), increased alkaline phosphatase (28%), decreased magnesium (27%), decreased potassium (26%), and decreased sodium (26%).
In the ASCENT study (locally advanced or metastatic triple-negative breast cancer), the most common adverse reactions (incidence ≥25%) were fatigue, diarrhea, nausea, alopecia, constipation, vomiting, abdominal pain, and decreased appetite. The most frequent serious adverse reactions (SAR) (>1%) were neutropenia (7%), diarrhea (4%), and pneumonia (3%). SAR were reported in 27% of patients, and 5% discontinued therapy due to adverse reactions. The most common Grade 3-4 lab abnormalities (incidence ≥25%) in the ASCENT study were reduced neutrophils, leukocytes, and lymphocytes.
In the TROPiCS-02 study (locally advanced or metastatic HR-positive, HER2-negative breast cancer), the most common adverse reactions (incidence ≥25%) were diarrhea, fatigue, nausea, alopecia, and constipation. The most frequent serious adverse reactions (SAR) (>1%) were diarrhea (5%), febrile neutropenia (4%), neutropenia (3%), abdominal pain, colitis, neutropenic colitis, pneumonia, and vomiting (each 2%). SAR were reported in 28% of patients, and 6% discontinued therapy due to adverse reactions. The most common Grade 3-4 lab abnormalities (incidence ≥25%) in the TROPiCS-02 study were reduced neutrophils and leukocytes.
Drug Interactions
UGT1A1 Inhibitors: Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38. Avoid administering UGT1A1 inhibitors with TRODELVY.
UGT1A1 Inducers: Exposure to SN-38 may be reduced in patients concomitantly receiving UGT1A1 enzyme inducers. Avoid administering UGT1A1 inducers with TRODELVY.
Please see full Prescribing Information, including BOXED WARNING.