For the treatment of adult patients with unresectable locally advanced or metastatic HR+/HER2- (IHC 0, IHC 1+, or IHC 2+/ISH-) breast cancer who have received endocrine-based therapy and at least 2 additional systemic therapies in the metastatic setting
TRODELVY has a manageable safety profile1,2,*
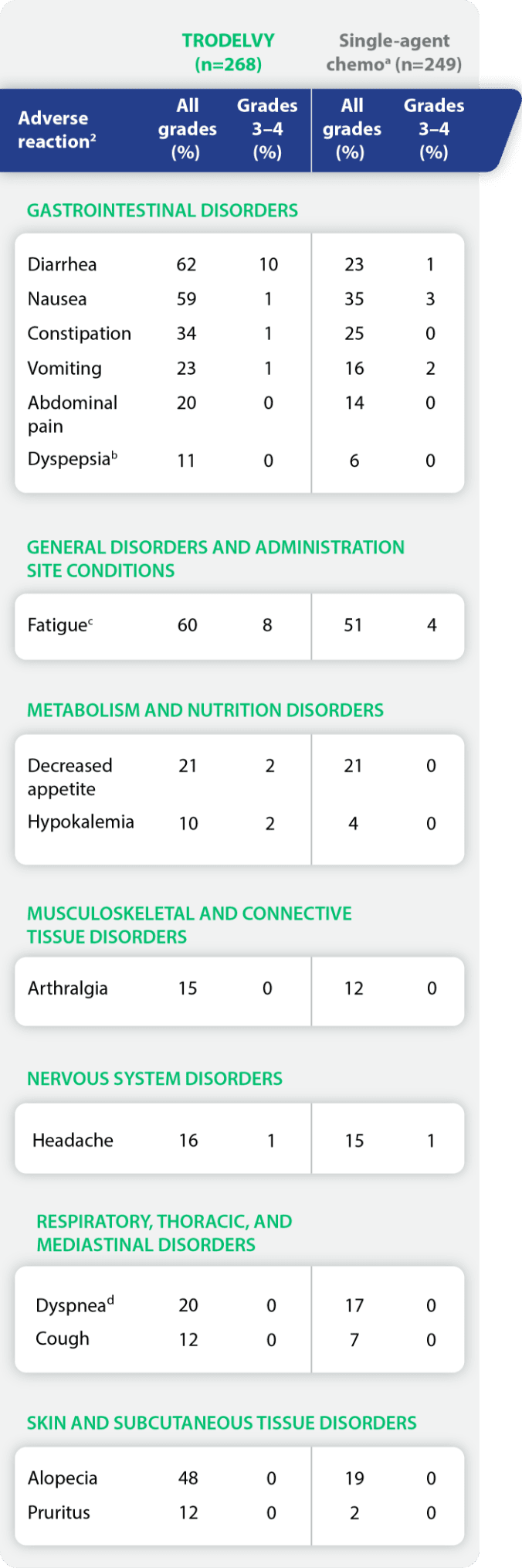
Adverse reactions in ≥10% of patients with HR+/HER2- mBC in TROPiCS-021
Graded per NCI CTCAE v.5.0.1
*The discontinuation rate in TROPiCS-02 was 6%, demonstrating that a majority of patients were able to remain on TRODELVY with proper management of adverse reactions.1
aSingle-agent chemotherapy included one of the following single agents: eribulin (n=130), vinorelbine (n=63), gemcitabine (n=56), or capecitabine (n=22).1
bIncluding dyspepsia, gastroesophageal reflux disease.1
cIncluding fatigue, asthenia.1
dIncluding dyspnea; dyspnea exertional.1
Other clinically significant adverse reactions (≤10%) in TROPiCS-02 included hypotension (5%), pain (5%), rhinorrhea (5%), hypocalcemia (3%), nasal congestion (3%), skin hyperpigmentation (3%), colitis or neutropenic colitis (2%), hyponatremia (2%), pneumonia (2%), proteinuria (1%), and enteritis (0.4%).1

Management of adverse reactions may require temporary interruption, dose reduction, or treatment discontinuation of TRODELVY as described in the tables below.1
| Dose level | Dosage and schedule |
|---|---|
| Recommended starting dose | 10 mg/kg once weekly on Days 1 and 8 of 21-day treatment cycles |
| First dose reduction | Reduce to 7.5 mg/kg |
| Second dose reduction | Reduce to 5 mg/kg |
| Requirement for further dose reduction | Permanently discontinue TRODELVY |
| Adverse reaction | Severity | Dose modification |
|---|---|---|
| Neutropenia | Grade 3-4 neutropenia (absolute neutrophil count [ANC] <1000/mm3) or febrile neutropenia |
|
| Nausea/Vomiting/ Diarrhea | Grade 3-4 nausea, vomiting, or diarrhea that is not controlled with antiemetics or anti-diarrheal agents |
|
| Infusion-Related Reaction | Grade 1-3 infusion-related reactions | Slow infusion rate or interrupt the infusion |
| Grade 4 infusion-related reactions | Discontinue TRODELVY | |
| Other Toxicities | Other Grade 3-4 toxicities of any duration despite optimal medical management |
|

Serious adverse reactions in TROPiCS-021
- Serious adverse reactions occurred in 28% of patients receiving TRODELVY
- Serious adverse reactions in >1% of patients receiving TRODELVY included diarrhea (5%), febrile neutropenia (4%), neutropenia (3%), abdominal pain, colitis, neutropenic colitis, pneumonia, and vomiting (each 2%)
Treatment discontinuation, reduction, and interruption in TROPiCS-021,2
- Similar rates of discontinuation due to adverse reactions: 6% with TRODELVY versus 4% with single-agent chemotherapy
- The most frequent (≥0.5%) adverse reactions leading to permanent discontinuation of TRODELVY were asthenia, general physical health deterioration, and neutropenia (each 0.7%)
- Adverse reactions leading to dose reduction occurred in 33% of patients treated with TRODELVY
- The most frequent (>5%) adverse reactions leading to dose reduction were neutropenia (16%) and diarrhea (8%)
- Adverse reactions leading to dose interruption occurred in 66% of patients treated with TRODELVY
- The most frequent (≥5%) adverse reaction leading to treatment interruption was neutropenia (50%)
- G-CSF was used in 54% of patients who received TRODELVY
Most common adverse reactions and lab abnormalities in TROPiCS-021
- The most common (≥25%) adverse reactions, including lab abnormalities, with TRODELVY were decreased leukocytes (88%), decreased neutrophils (83%), decreased hemoglobin (73%), decreased lymphocytes (65%), diarrhea (62%), fatigue (60%), nausea (59%), alopecia (48%), increased glucose (37%), constipation (34%), and decreased albumin (32%)

Chemo=chemotherapy; G-CSF=granulocyte-colony stimulating factor; HER2-=human epidermal growth factor receptor 2–negative; HR+=hormone receptor–positive; ILD=interstitial lung disease; mBC=metastatic breast cancer; NCI CTCAE=National Cancer Institute Common Terminology Criteria for Adverse Events; PI=prescribing information.
References: 1. TRODELVY. Prescribing Information. Gilead Sciences, Inc.; March 2025. 2. Rugo HS, Bardia A, Marmé F, et al. Sacituzumab govitecan in hormone receptor–positive/human epidermal growth factor receptor 2–negative metastatic breast cancer. J Clin Oncol. 2022;40(29):3365-3376.
TRODELVY® (sacituzumab govitecan-hziy) is a Trop-2-directed antibody and topoisomerase inhibitor conjugate indicated for the treatment of adult patients with unresectable locally advanced or metastatic hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting.
- TRODELVY can cause severe, life-threatening, or fatal neutropenia. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Primary prophylaxis with G-CSF is recommended for all patients at increased risk of febrile neutropenia. Initiate anti-infective treatment in patients with febrile neutropenia without delay.
- TRODELVY can cause severe diarrhea. Monitor patients with diarrhea and give fluid and electrolytes as needed. At the onset of diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤Grade 1 and reduce subsequent doses.
CONTRAINDICATIONS
- Severe hypersensitivity reaction to TRODELVY.
WARNINGS AND PRECAUTIONS
Neutropenia: Severe, life-threatening, or fatal neutropenia can occur as early as the first cycle of treatment and may require dose modification. Neutropenia occurred in 64% of patients treated with TRODELVY. Grade 3-4 neutropenia occurred in 49% of patients. Febrile neutropenia occurred in 6%. Neutropenic colitis occurred in 1.4%. Primary prophylaxis with G-CSF is recommended starting in the first cycle of treatment in all patients at increased risk of febrile neutropenia, including older patients, patients with previous neutropenia, poor performance status, organ dysfunction, or multiple comorbidities. Monitor absolute neutrophil count (ANC) during treatment. Withhold TRODELVY for ANC below 1500/mm3 on Day 1 of any cycle or below 1000/mm3 on Day 8 of any cycle. Withhold TRODELVY for neutropenic fever. Treat neutropenia with G-CSF and administer prophylaxis in subsequent cycles as clinically indicated or indicated in Table 2 of USPI.
Diarrhea: Diarrhea occurred in 64% of all patients treated with TRODELVY. Grade 3-4 diarrhea occurred in 11% of patients. One patient had intestinal perforation following diarrhea. Diarrhea that led to dehydration and subsequent acute kidney injury occurred in 0.7% of all patients. Withhold TRODELVY for Grade 3-4 diarrhea and resume when resolved to ≤Grade 1. At onset, evaluate for infectious causes and if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (e.g., fluid and electrolyte substitution) may also be employed as clinically indicated. Patients who exhibit an excessive cholinergic response to treatment can receive appropriate premedication (e.g., atropine) for subsequent treatments.
Hypersensitivity and Infusion-Related Reactions: TRODELVY can cause serious hypersensitivity reactions including life-threatening anaphylactic reactions. Severe signs and symptoms included cardiac arrest, hypotension, wheezing, angioedema, swelling, pneumonitis, and skin reactions. Hypersensitivity reactions within 24 hours of dosing occurred in 35% of patients. Grade 3-4 hypersensitivity occurred in 2% of patients. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.2%. The incidence of anaphylactic reactions was 0.2%. Pre-infusion medication is recommended. Have medications and emergency equipment to treat such reactions available for immediate use. Observe patients closely for hypersensitivity and infusion-related reactions during each infusion and for at least 30 minutes after completion of each infusion. Permanently discontinue TRODELVY for Grade 4 infusion-related reactions.
Nausea and Vomiting: TRODELVY is emetogenic and can cause severe nausea and vomiting. Nausea occurred in 64% of all patients treated with TRODELVY and Grade 3-4 nausea occurred in 3% of these patients. Vomiting occurred in 35% of patients and Grade 3-4 vomiting occurred in 2% of these patients. Premedicate with a two or three drug combination regimen (e.g., dexamethasone with either a 5-HT3 receptor antagonist or an NK1 receptor antagonist as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting (CINV). Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting and resume with additional supportive measures when resolved to Grade ≤1. Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity: Patients homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia and may be at increased risk for other adverse reactions with TRODELVY. The incidence of Grade 3-4 neutropenia was 58% in patients homozygous for the UGT1A1*28, 49% in patients heterozygous for the UGT1A1*28 allele, and 43% in patients homozygous for the wild-type allele. The incidence of Grade 3-4 anemia was 21% in patients homozygous for the UGT1A1*28 allele, 10% in patients heterozygous for the UGT1A1*28 allele, and 9% in patients homozygous for the wild-type allele. Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on clinical assessment of the onset, duration and severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 function.
Embryo-Fetal Toxicity: Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
ADVERSE REACTIONS
In the pooled safety population, the most common (≥25%) adverse reactions including laboratory abnormalities were decreased leukocyte count (84%), decreased neutrophil count (75%), decreased hemoglobin (69%), diarrhea (64%), nausea (64%), decreased lymphocyte count (63%), fatigue (51%), alopecia (45%), constipation (37%), increased glucose (37%), decreased albumin (35%), vomiting (35%), decreased appetite (30%), decreased creatinine clearance (28%), increased alkaline phosphatase (28%), decreased magnesium (27%), decreased potassium (26%), and decreased sodium (26%).
In the TROPiCS-02 study, the most common adverse reactions (incidence ≥25%) were diarrhea, fatigue, nausea, alopecia, and constipation. The most frequent serious adverse reactions (SAR) (>1%) were diarrhea (5%), febrile neutropenia (4%), neutropenia (3%), abdominal pain, colitis, neutropenic colitis, pneumonia, and vomiting (each 2%). SAR were reported in 28% of patients, and 6% discontinued therapy due to adverse reactions. The most common Grade 3-4 lab abnormalities (incidence ≥25%) in the TROPiCS-02 study were reduced neutrophils and leukocytes.
DRUG INTERACTIONS
UGT1A1 Inhibitors: Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38. Avoid administering UGT1A1 inhibitors with TRODELVY.
UGT1A1 Inducers: Exposure to SN-38 may be reduced in patients concomitantly receiving UGT1A1 enzyme inducers. Avoid administering UGT1A1 inducers with TRODELVY.
Please see full Prescribing Information, including BOXED WARNING.