TRODELVY in 2L+ mTNBC
TRODELVY, a proven standard-of-care option in mTNBC
NCCN Guidelines®
NCCN Category 1 preferred option for 2L mTNBC2
SURVIVAL DATA
ASCENT Study Design: ASCENT was a Phase 3, randomized, active-controlled, open-label study (N=529) in patients with unresectable locally advanced or mTNBC who had relapsed after at least 2 prior chemotherapies, at least one of them for metastatic disease. The efficacy analysis included PFS in brain-met–negative patients by BICR based on RECIST 1.1 criteria (primary endpoint), with PFS for the full population (all patients with and without brain metastases) and OS as secondary endpoints. Patients were randomized (1:1) to receive TRODELVY 10 mg/kg as an IV infusion on Days 1 and 8 of a 21-day cycle (n=267) or physician's choice of single-agent chemotherapy (n=262), which included eribulin, vinorelbine, gemcitabine, or capecitabine. Patients were treated until disease progression or unacceptable toxicity. 88% of patients in the full population were brain-met–negative.1,3
A Phase 3, randomized, active-controlled, open-label study
Scroll left to review

Single-agent chemotherapy was determined by the investigator before randomization from one of the following choices: eribulin (n=139), capecitabine (n=33), gemcitabine (n=38), or vinorelbine (n=52).1
Patients with brain metastases were allowed to enroll up to a predefined maximum of 15% of patients in the ASCENT study; MRI was required prior to enrollment for patients with known or suspected brain metastases. Patients with known Gilbert’s disease or bone-only disease were excluded.1
aAll patients received previous taxane treatment in either the adjuvant, neoadjuvant, or advanced stage unless there was a contraindication or intolerance to taxanes during or at the end of the first taxane cycle.1
The population of ASCENT has characteristics that may resemble those of your patients1
DEMOGRAPHICS1 |
|
|---|---|
| Median age: | 54 years (range: 27-82 years); 81% <65 years |
| Sex: | 99.6% female |
| Race/ethnicity: | 79% White; 12% Black/African American |
DISEASE CHARACTERISTICS |
|
|---|---|
| Hepatic metastases (visceral disease): | 42% |
| Brain metastases: | 12% |
| BRCA1/BRCA2 positive: | 9% |
| ECOG performance status: | 0 (43%); 1 (57%) |
TREATMENT HISTORY1,2 |
|
|---|---|
| Prior PD-1/PD-L1 therapy: | 29% |
| 1 prior line of systemic therapy in metastatic settingb: | 13% in TRODELVY group |
bIn addition to having disease recurrence or progression within 12 months of neoadjuvant/adjuvant systemic therapy.1
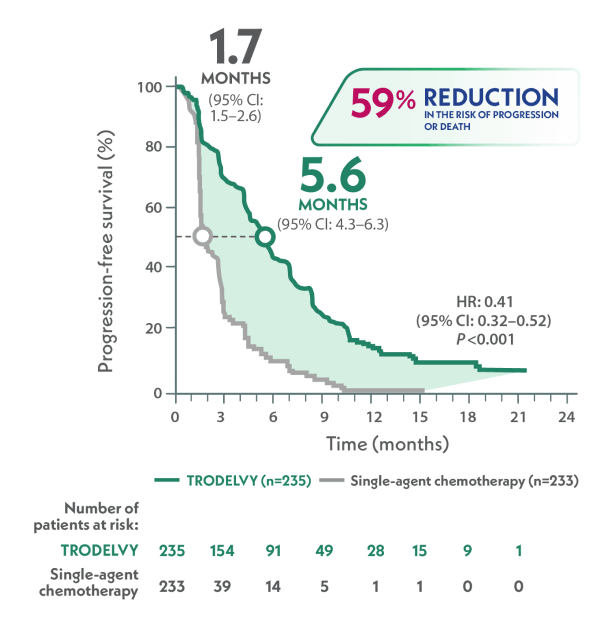
More than 3x as long median PFS with TRODELVY compared to single-agent chemotherapy in 2L+ mTNBC1,3
Primary endpoint: mPFS in the brain-met–negative population by BICR per RECIST 1.1 criteria1,a
>3x AS LONG
VS
HR: 0.41 (95% CI: 0.32–0.52); P<0.001
- 88% of patients in the ASCENT study were brain-met–negative
- TRODELVY was studied in patients across IHC status (IHC 0, IHC 1+, and IHC 2+/ISH–)4
aPFS was defined as the time from the date of randomization to the date of the first radiological disease progression or death due to any cause, whichever came first.1
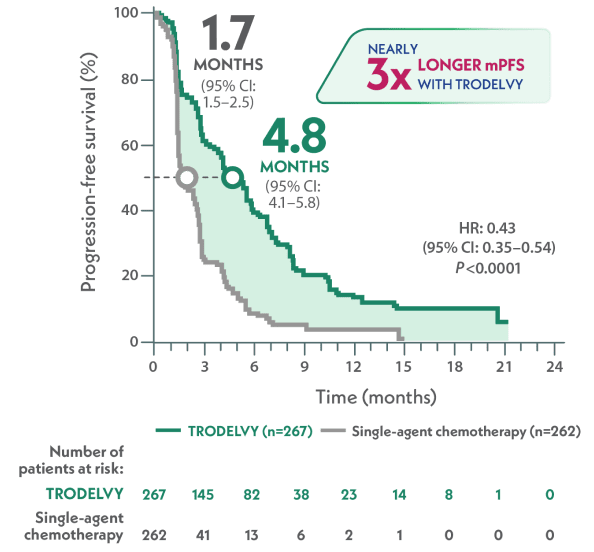
Nearly 3x as long median PFS with TRODELVY compared to single-agent chemotherapy1,3
mPFS in the full population by BICR per RECIST 1.1 criteria1,a
~3x LONGER
VS
HR: 0.43 (95% CI: 0.35–0.54); P<0.0001
aPFS was defined as the time from the date of randomization to the date of the first radiological disease progression or death due to any cause, whichever came first.1
Explore PFS data in 1L mTNBC across PD-L1 status
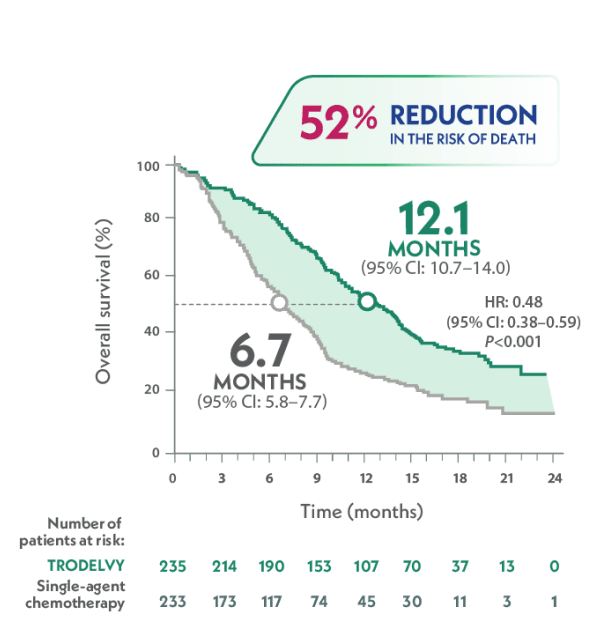
Statistically significant mOS benefit vs single-agent chemotherapy in 2L+ mTNBC1,3
Nearly 2x as long median OS with TRODELVY compared to single-agent chemotherapy3
Secondary endpoint: mOS in the brain-met–negative population3
NEARLY 2x AS LONG
VS
HR: 0.48 (95% CI: 0.38–0.59); P<0.001
mOS in the full population of ASCENT was 11.8 months with TRODELVY (95% CI: 10.5–13.8) vs 6.9 months with chemotherapy (95% CI: 5.9–7.6) (n=262); HR: 0.51 (95% CI: 0.41–0.62); P<0.00011
- 88% of patients in the ASCENT study were brain-met–negative1
- TRODELVY was studied in patients across IHC status (IHC 0, IHC 1+, and IHC 2+/ISH–)4
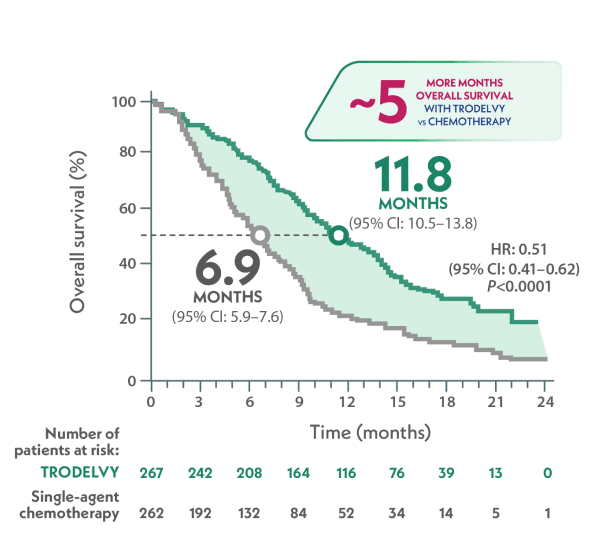
Nearly 5 months longer median OS with TRODELVY compared to single-agent chemotherapy1
mOS in the full population1
~5 MONTHS LONGER
VS
HR: 0.51 (95% CI: 0.41-0.62); P<0.0001
- Similar to how patients may present in a clinical setting, the full population in ASCENT included patients both with stable brain metastases (12%) and without them (88%)1
REAL-WORLD OUTCOMES
Real-world results support the proven efficacy of TRODELVY seen in the Phase 3 ASCENT trial5
A retrospective, observational cohort study assessed real-world clinical outcomes in patients with mTNBC treated with TRODELVY as 2L and later.5
Limitations: These EHR-derived, retrospective data were not powered for statistical analysis and should be considered descriptive only. The results require cautious interpretation and could represent chance findings. Data entry errors could have occurred. It is likely there are missing data.5
Real-world mPFS and mOS in 2L and 3L+ mTNBC5
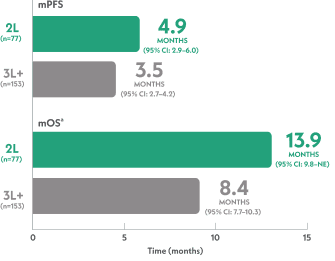
aFrom index data.5
- For the total population (N=230), the mPFS was 3.8 months (95% CI: 3.1–4.3) and the mOS was 10 months (95% CI: 8.3–11.1)5
Numerically greater outcomes were seen in 2L vs 3L+ in a real-world treatment setting5
Real-world OS rate (95% CI)
| 2L mTNBC (n=77) | 3L+ mTNBC (n=153) | Total (N=230) | |
|
|
51% (37–64) | 35% (26–44) | 40% (33–48) |
|
|
32% (13–54) | 20% (11–29) | 23% (15–32) |
| 2L mTNBC (n=77) |
3L+ mTNBC (n=153) |
Total (N=230) |
|
|
||
| 51% (37–64) |
35% (26–44) |
40% (33–48) |
|
|
||
| 32% (13–54) |
20% (11–29) |
23% (15–32) |
Real-world safety data (n=230)5
Limitations: Underreporting of AEs and dose modifications in physicians’ notes may have occurred with an unknown impact on the results.5
- Dose reduction and interruptions were observed in 34% and 58% of all patients, with 26% and 39% due to toxicity, respectively; 7% discontinued TRODELVY due to toxicity5
- Fatigue was reported in 45%, neutropenia in 33%, and diarrhea in 30% of patients5
- Concomitant administration of G-CSF was observed in 58% of all patients, with most of these patients (99/134=74%) having received G-CSF with prior anticancer treatment5
- 15% of patients received G-CSF for the first time during treatment with TRODELVY. Among these patients, the median time from TRODELVY start to G-CSF use was 8.5 days (IQR: 8.0–29.0)5
Methods for the real-world study5
A retrospective, observational cohort study used ConcertAI database de-identified EHR data, with safety data supplemented with physicians’ notes. From April 2020 to May 2022, 230 adult female patients with mTNBC were treated with TRODELVY in the 2L or later setting. 33% and 67% of patients received TRODELVY in the 2L and 3L+ settings, respectively. The median starting dose was 10 mg/kg, with a median of 9 doses. The median treatment duration was 3.8 months among all patients and 4.2 months among 2L patients. The maximum treatment duration among all patients was 25.8 months. Data included in this analysisa were cut off in August 2022. At the end of the study period, 21 (9%) patients were still receiving TRODELVY.a
aData cutoff to allow for ≥3-month data accrual. Sensitivity analysis was performed using ≥6-month data accrual.5
Across indications, TRODELVY has a well-characterized safety profile
2L=second line; 3L=third line; ADC=antibody-drug conjugate; AE=adverse event; BICR=blinded independent central review; CI=confidence interval; EHR=electronic health records; G-CSF=granulocyte colony-stimulating factor; HER2=human epidermal growth factor receptor 2; HR=hazard ratio; IHC=immunohistochemistry; IV=intravenous; mOS=median OS; mPFS=median PFS; mTNBC=metastatic triple-negative breast cancer; NCCN=National Comprehensive Cancer Network; OS=overall survival; PFS=progression-free survival; RECIST=response evaluation criteria in solid tumors; Trop-2=trophoblast cell-surface antigen-2.
References: 1. TRODELVY. Prescribing Information. Gilead Sciences, Inc.; 2026. 2. Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Breast Cancer V.2.2026. © National Comprehensive Cancer Network, Inc. 2026. All rights reserved. Accessed March 9, 2026. To view the most recent and complete version of the guideline, go online to NCCN.org. 3. Bardia A, Hurvitz SA, Tolaney SM, et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N Engl J Med. 2021;384(16):1529-1541. 4. Immunomedics, Inc. An international, multi-center, open-label, randomized, phase III trial of sacituzumab govitecan versus treatment of physician choice in patients with metastatic triple-negative breast cancer who received at least two prior treatments. Published November 18, 2015. Updated June 22, 2017. Accessed October 10, 2025. https://www.nejm.org/doi/suppl/l0.1056/NEJMoa2028485/suppl_file/nejmoa2028485_protocol.pdf 5. Kalinsky K, Spring L, Yam C, et al. Real-world use patterns, effectiveness, and tolerability of sacituzumab govitecan for second-line and later-line treatment of metastatic triple-negative breast cancer in the United States. Breast Cancer Res Treat. 2024;208(1):203-214.
TRODELVY® (sacituzumab govitecan-hziy) is a Trop-2–directed antibody and topoisomerase inhibitor conjugate indicated in adult patients:
Locally Advanced or Metastatic Triple-Negative Breast Cancer
First Line
- As a single agent for the first-line treatment of unresectable locally advanced or metastatic triple-negative breast cancer (mTNBC) who are not candidates for PD-1 or PD-L1 inhibitor-based therapy
- In combination with pembrolizumab or pembrolizumab and berahyaluronidase alfa-pmph for the first-line treatment of unresectable locally advanced or mTNBC whose tumors express PD-L1 [Combined Positive Score (CPS ≥10)] as determined by an FDA-authorized test
Second Line or Later
- For the treatment of unresectable locally advanced or mTNBC who have received two or more prior systemic therapies, at least one of them for metastatic disease.
Locally Advanced or Metastatic HR-positive, HER2-negative Breast Cancer
- For the treatment of unresectable locally advanced or metastatic hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+, or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting.
Important Safety Information
Tap for Important Safety Information, including BOXED WARNING: Neutropenia and Diarrhea.
Boxed Warning: neutropenia and diarrhea
- TRODELVY can cause severe, life-threatening, or fatal neutropenia. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Primary prophylaxis with G-CSF is recommended for all patients at increased risk of febrile neutropenia. Initiate anti-infective treatment in patients with febrile neutropenia without delay.
- TRODELVY can cause severe diarrhea. Monitor patients with diarrhea and give fluid and electrolytes as needed. At the onset of diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤Grade 1 and reduce subsequent doses.
Contraindications
- Severe hypersensitivity reaction to TRODELVY.
Warnings and precautions
Neutropenia: Severe, life-threatening, or fatal neutropenia can occur as early as the first cycle of treatment and may require dose modification. Neutropenia occurred in 64% of patients treated with TRODELVY. Grade 3-4 neutropenia occurred in 48% of patients. Febrile neutropenia occurred in 6%. Neutropenic colitis occurred in 1.4%. Primary prophylaxis with G-CSF is recommended starting in the first cycle of treatment in all patients at increased risk of febrile neutropenia, including older patients, patients with previous neutropenia, poor performance status, organ dysfunction, or multiple comorbidities. Monitor absolute neutrophil count (ANC) during treatment. Withhold TRODELVY for ANC below 1500/mm3 on Day 1 of any cycle or below 1000/mm3 on Day 8 of any cycle. Withhold TRODELVY for neutropenic fever. Treat neutropenia with G-CSF and administer prophylaxis in subsequent cycles as clinically indicated or indicated in Table 2 of USPI.
Diarrhea: Diarrhea occurred in 62% of all patients treated with TRODELVY. Grade 3-4 diarrhea occurred in 10% of patients. One patient had intestinal perforation following diarrhea. Diarrhea that led to dehydration and subsequent acute kidney injury occurred in 0.6% of all patients. Withhold TRODELVY for Grade 3-4 diarrhea and resume when resolved to ≤Grade 1. At onset, evaluate for infectious causes and, if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (eg, fluid and electrolyte replacement) may also be employed as clinically indicated. Patients who exhibit an excessive cholinergic response to treatment can receive appropriate premedication (eg, atropine) for subsequent treatments.
Hypersensitivity and Infusion-Related Reactions: TRODELVY can cause serious hypersensitivity reactions, including life-threatening anaphylactic reactions. Severe signs and symptoms included cardiac arrest, hypotension, wheezing, angioedema, swelling, and skin reactions. Hypersensitivity reactions occurred in 28% of patients with 13% occurring within 24 hours of dosage. Grade 3-4 hypersensitivity occurred in 1.5% of patients with 0.4% of these occurring within 24 hours of dosage. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.4%. The incidence of anaphylactic reaction was <0.1%. Pre-infusion medication is recommended. Have medications and emergency equipment to treat such reactions available for immediate use. Closely monitor patients for hypersensitivity and infusion-related reactions during each infusion and for at least 30 minutes after completion of each infusion. Permanently discontinue TRODELVY for Grade 4 infusion-related reactions.
Nausea and Vomiting: TRODELVY is emetogenic and can cause severe nausea and vomiting. Nausea occurred in 63% of all patients treated with TRODELVY, and Grade 3-4 nausea occurred in 3% of these patients. Vomiting occurred in 33% of patients, and Grade 3-4 vomiting occurred in 2% of these patients. Premedicate with a two- or three-drug combination regimen (eg, dexamethasone with either a 5-HT3 receptor antagonist or an NK1 receptor antagonist, as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting. Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting and resume with additional supportive measures when resolved to ≤Grade 1. Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity: Patients homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia and may be at increased risk for other adverse reactions with TRODELVY. The incidence of Grade 3-4 neutropenia was 57% in patients homozygous for the UGT1A1*28 allele, 48% in patients heterozygous for the UGT1A1*28 allele, and 41% in patients homozygous for the wild-type allele. The incidence of Grade 3-4 anemia was 17% in patients homozygous for the UGT1A1*28 allele, 9% in patients heterozygous for the UGT1A1*28 allele, and 8% in patients homozygous for the wild-type allele. Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on clinical assessment of the onset, duration, and severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 function.
Embryo-Fetal Toxicity: Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
Adverse Reactions
In the pooled safety population of TRODELVY as a single agent, the most common (≥25%) adverse reactions, including laboratory abnormalities, were decreased leukocyte count (83%), decreased neutrophil count (77%), decreased hemoglobin (71%), nausea (63%), diarrhea (62%), decreased lymphocyte count (60%), fatigue (59%), alopecia (47%), increased glucose (40%), constipation (37%), vomiting (33%), decreased albumin (32%), increased alkaline phosphatase (30%), decreased appetite (28%), abdominal pain (27%), decreased creatinine clearance (27%), decreased magnesium and potassium (26% each).
In the safety population of TRODELVY in combination with pembrolizumab, the most common (≥25%) adverse reactions, including laboratory abnormalities, were decreased neutrophil count and hemoglobin (86% each), decreased leukocyte count (84%), diarrhea (72%), nausea (68%), decreased lymphocyte count (61%), fatigue (58%), alopecia (52%), increased alkaline phosphatase and glucose (50% each), increased alanine aminotransferase (47%), constipation (41%), increased aspartate aminotransferase (40%), rash (37%), decreased potassium (35%), increased lactate dehydrogenase (34%), vomiting (29%), abdominal pain, headache, and increased eosinophils (26% each), and decreased albumin (25%).
In the ASCENT-03 study (single agent in previously untreated, unresectable locally advanced or mTNBC), the most common adverse reactions (incidence ≥25%) were nausea, diarrhea, alopecia, fatigue, constipation, and vomiting. The most frequent serious adverse reactions (SAR) (>2%) were diarrhea, febrile neutropenia, and neutropenia (3.6% each), and pneumonia (2.9%). SAR occurred in 26% of patients, and 3.6% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 2.5% of patients and included sepsis (1.1%), and acute respiratory failure, neutropenic colitis, pneumonia, and septic shock (0.4% each). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils and leukocytes.
In the ASCENT-04 study (in combination with pembrolizumab in previously untreated, unresectable locally advanced or mTNBC whose tumors express PD-L1), the most common adverse reactions (incidence ≥25%) were diarrhea, nausea, fatigue, alopecia, constipation, rash, vomiting, abdominal pain, and headache. The most frequent SAR (≥2%) were febrile neutropenia (7%), neutropenia (6%), diarrhea (5%), and fatigue and pneumonia (2.3% each). SAR occurred in 38% of patients, and 7% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 3.2% of patients and included death (unknown cause) (0.9%) and completed suicide, neutropenic sepsis, sepsis, pneumonia, and pulmonary embolism (0.5% each). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils and leukocytes.
In the ASCENT study (previously treated locally advanced or mTNBC), the most common adverse reactions (incidence ≥25%) were fatigue, diarrhea, nausea, alopecia, constipation, vomiting, abdominal pain, and decreased appetite. The most frequent SAR (>1%) were neutropenia (7%), diarrhea (4%), and pneumonia (3%). SAR occurred in 27% of patients, and 5% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 1.2% of patients and included respiratory failure (0.8%) and pneumonia (0.4%). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils, leukocytes, and lymphocytes.
In the TROPiCS-02 study (locally advanced or metastatic HR+/HER2– breast cancer), the most common adverse reactions (incidence ≥25%) were diarrhea, fatigue, nausea, alopecia, and constipation. The most frequent SAR (>1%) were diarrhea (5%), febrile neutropenia (4.1%), neutropenia (3%), abdominal pain (2.2%), neutropenic colitis and vomiting (1.9% each), and colitis and pneumonia (1.5% each). SAR occurred in 28% of patients, and 6% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 2.2% of patients and included arrhythmia, COVID-19 pneumonia, pneumonia, nervous system disorder, pulmonary embolism, and septic shock (0.4% each). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils and leukocytes.
Drug Interactions
UGT1A1 Inhibitors: Avoid administering UGT1A1 inhibitors with TRODELVY. SN-38 is a UGT1A1 substrate. Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38.
UGT1A1 Inducers: Avoid administering UGT1A1 inducers with TRODELVY. SN-38 is a UGT1A1 substrate. Concomitant administration of TRODELVY with inducers of UGT1A1 may reduce exposure to SN-38.
Please see full Prescribing Information, including BOXED WARNING.

TRODELVY® (sacituzumab govitecan-hziy) is a Trop-2–directed antibody and topoisomerase inhibitor conjugate indicated in adult patients:
Locally Advanced or Metastatic Triple-Negative Breast Cancer
First Line
- As a single agent for the first-line treatment of unresectable locally advanced or metastatic triple-negative breast cancer (mTNBC) who are not candidates for PD-1 or PD-L1 inhibitor-based therapy
- In combination with pembrolizumab or pembrolizumab and berahyaluronidase alfa-pmph for the first-line treatment of unresectable locally advanced or mTNBC whose tumors express PD-L1 [Combined Positive Score (CPS ≥10)] as determined by an FDA-authorized test
Second Line or Later
- For the treatment of unresectable locally advanced or mTNBC who have received two or more prior systemic therapies, at least one of them for metastatic disease.
Locally Advanced or Metastatic HR-positive, HER2-negative Breast Cancer
- For the treatment of unresectable locally advanced or metastatic hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+, or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting.
Important Safety Information
Tap for Important Safety Information, including BOXED WARNING: Neutropenia and Diarrhea.
Boxed Warning: neutropenia and diarrhea
- TRODELVY can cause severe, life-threatening, or fatal neutropenia. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Primary prophylaxis with G-CSF is recommended for all patients at increased risk of febrile neutropenia. Initiate anti-infective treatment in patients with febrile neutropenia without delay.
- TRODELVY can cause severe diarrhea. Monitor patients with diarrhea and give fluid and electrolytes as needed. At the onset of diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤Grade 1 and reduce subsequent doses.
Contraindications
- Severe hypersensitivity reaction to TRODELVY.
Warnings and precautions
Neutropenia: Severe, life-threatening, or fatal neutropenia can occur as early as the first cycle of treatment and may require dose modification. Neutropenia occurred in 64% of patients treated with TRODELVY. Grade 3-4 neutropenia occurred in 48% of patients. Febrile neutropenia occurred in 6%. Neutropenic colitis occurred in 1.4%. Primary prophylaxis with G-CSF is recommended starting in the first cycle of treatment in all patients at increased risk of febrile neutropenia, including older patients, patients with previous neutropenia, poor performance status, organ dysfunction, or multiple comorbidities. Monitor absolute neutrophil count (ANC) during treatment. Withhold TRODELVY for ANC below 1500/mm3 on Day 1 of any cycle or below 1000/mm3 on Day 8 of any cycle. Withhold TRODELVY for neutropenic fever. Treat neutropenia with G-CSF and administer prophylaxis in subsequent cycles as clinically indicated or indicated in Table 2 of USPI.
Diarrhea: Diarrhea occurred in 62% of all patients treated with TRODELVY. Grade 3-4 diarrhea occurred in 10% of patients. One patient had intestinal perforation following diarrhea. Diarrhea that led to dehydration and subsequent acute kidney injury occurred in 0.6% of all patients. Withhold TRODELVY for Grade 3-4 diarrhea and resume when resolved to ≤Grade 1. At onset, evaluate for infectious causes and, if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (eg, fluid and electrolyte replacement) may also be employed as clinically indicated. Patients who exhibit an excessive cholinergic response to treatment can receive appropriate premedication (eg, atropine) for subsequent treatments.
Hypersensitivity and Infusion-Related Reactions: TRODELVY can cause serious hypersensitivity reactions, including life-threatening anaphylactic reactions. Severe signs and symptoms included cardiac arrest, hypotension, wheezing, angioedema, swelling, and skin reactions. Hypersensitivity reactions occurred in 28% of patients with 13% occurring within 24 hours of dosage. Grade 3-4 hypersensitivity occurred in 1.5% of patients with 0.4% of these occurring within 24 hours of dosage. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.4%. The incidence of anaphylactic reaction was <0.1%. Pre-infusion medication is recommended. Have medications and emergency equipment to treat such reactions available for immediate use. Closely monitor patients for hypersensitivity and infusion-related reactions during each infusion and for at least 30 minutes after completion of each infusion. Permanently discontinue TRODELVY for Grade 4 infusion-related reactions.
Nausea and Vomiting: TRODELVY is emetogenic and can cause severe nausea and vomiting. Nausea occurred in 63% of all patients treated with TRODELVY, and Grade 3-4 nausea occurred in 3% of these patients. Vomiting occurred in 33% of patients, and Grade 3-4 vomiting occurred in 2% of these patients. Premedicate with a two- or three-drug combination regimen (eg, dexamethasone with either a 5-HT3 receptor antagonist or an NK1 receptor antagonist, as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting. Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting and resume with additional supportive measures when resolved to ≤Grade 1. Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity: Patients homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia and may be at increased risk for other adverse reactions with TRODELVY. The incidence of Grade 3-4 neutropenia was 57% in patients homozygous for the UGT1A1*28 allele, 48% in patients heterozygous for the UGT1A1*28 allele, and 41% in patients homozygous for the wild-type allele. The incidence of Grade 3-4 anemia was 17% in patients homozygous for the UGT1A1*28 allele, 9% in patients heterozygous for the UGT1A1*28 allele, and 8% in patients homozygous for the wild-type allele. Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on clinical assessment of the onset, duration, and severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 function.
Embryo-Fetal Toxicity: Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
Adverse Reactions
In the pooled safety population of TRODELVY as a single agent, the most common (≥25%) adverse reactions, including laboratory abnormalities, were decreased leukocyte count (83%), decreased neutrophil count (77%), decreased hemoglobin (71%), nausea (63%), diarrhea (62%), decreased lymphocyte count (60%), fatigue (59%), alopecia (47%), increased glucose (40%), constipation (37%), vomiting (33%), decreased albumin (32%), increased alkaline phosphatase (30%), decreased appetite (28%), abdominal pain (27%), decreased creatinine clearance (27%), decreased magnesium and potassium (26% each).
In the safety population of TRODELVY in combination with pembrolizumab, the most common (≥25%) adverse reactions, including laboratory abnormalities, were decreased neutrophil count and hemoglobin (86% each), decreased leukocyte count (84%), diarrhea (72%), nausea (68%), decreased lymphocyte count (61%), fatigue (58%), alopecia (52%), increased alkaline phosphatase and glucose (50% each), increased alanine aminotransferase (47%), constipation (41%), increased aspartate aminotransferase (40%), rash (37%), decreased potassium (35%), increased lactate dehydrogenase (34%), vomiting (29%), abdominal pain, headache, and increased eosinophils (26% each), and decreased albumin (25%).
In the ASCENT-03 study (single agent in previously untreated, unresectable locally advanced or mTNBC), the most common adverse reactions (incidence ≥25%) were nausea, diarrhea, alopecia, fatigue, constipation, and vomiting. The most frequent serious adverse reactions (SAR) (>2%) were diarrhea, febrile neutropenia, and neutropenia (3.6% each), and pneumonia (2.9%). SAR occurred in 26% of patients, and 3.6% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 2.5% of patients and included sepsis (1.1%), and acute respiratory failure, neutropenic colitis, pneumonia, and septic shock (0.4% each). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils and leukocytes.
In the ASCENT-04 study (in combination with pembrolizumab in previously untreated, unresectable locally advanced or mTNBC whose tumors express PD-L1), the most common adverse reactions (incidence ≥25%) were diarrhea, nausea, fatigue, alopecia, constipation, rash, vomiting, abdominal pain, and headache. The most frequent SAR (≥2%) were febrile neutropenia (7%), neutropenia (6%), diarrhea (5%), and fatigue and pneumonia (2.3% each). SAR occurred in 38% of patients, and 7% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 3.2% of patients and included death (unknown cause) (0.9%) and completed suicide, neutropenic sepsis, sepsis, pneumonia, and pulmonary embolism (0.5% each). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils and leukocytes.
In the ASCENT study (previously treated locally advanced or mTNBC), the most common adverse reactions (incidence ≥25%) were fatigue, diarrhea, nausea, alopecia, constipation, vomiting, abdominal pain, and decreased appetite. The most frequent SAR (>1%) were neutropenia (7%), diarrhea (4%), and pneumonia (3%). SAR occurred in 27% of patients, and 5% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 1.2% of patients and included respiratory failure (0.8%) and pneumonia (0.4%). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils, leukocytes, and lymphocytes.
In the TROPiCS-02 study (locally advanced or metastatic HR+/HER2– breast cancer), the most common adverse reactions (incidence ≥25%) were diarrhea, fatigue, nausea, alopecia, and constipation. The most frequent SAR (>1%) were diarrhea (5%), febrile neutropenia (4.1%), neutropenia (3%), abdominal pain (2.2%), neutropenic colitis and vomiting (1.9% each), and colitis and pneumonia (1.5% each). SAR occurred in 28% of patients, and 6% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 2.2% of patients and included arrhythmia, COVID-19 pneumonia, pneumonia, nervous system disorder, pulmonary embolism, and septic shock (0.4% each). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils and leukocytes.
Drug Interactions
UGT1A1 Inhibitors: Avoid administering UGT1A1 inhibitors with TRODELVY. SN-38 is a UGT1A1 substrate. Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38.
UGT1A1 Inducers: Avoid administering UGT1A1 inducers with TRODELVY. SN-38 is a UGT1A1 substrate. Concomitant administration of TRODELVY with inducers of UGT1A1 may reduce exposure to SN-38.
Please see full Prescribing Information, including BOXED WARNING.