Scroll left to select an efficacy measure
Progression-Free Survival in Pretreated HR+/HER2– mBC
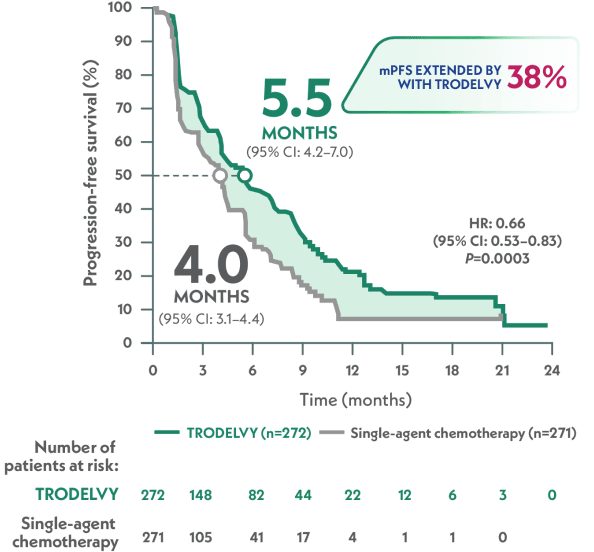
Statistically significant and clinically meaningful mPFS benefit1
MEDIAN PROGRESSION-FREE SURVIVAL
TROPiCS-02 evaluated outcomes with TRODELVY compared with chemotherapy1
TRODELVY was studied in TROPiCS-02—a randomized, active-controlled, open-label, Phase 3 study (N=543) of patients with HR+/HER2– mBC who were previously treated with ≥1 endocrine therapy, a CDK4/6 inhibitor, and a taxane in any setting, and who had received 2 to 4 lines of chemotherapy in the metastatic setting. The primary endpoint was progression-free survival (PFS) as assessed by BICR per the RECIST v1.1 criteria for TRODELVY compared to single-agent chemotherapy of investigator’s choice (eribulin, vinorelbine, gemcitabine, or capecitabine). Secondary endpoints included overall survival (OS), as well as assessment of safety and quality-of-life measures.1,2
A Phase 3, randomized, active-controlled, open-label study1-3:
Scroll left to review

Single-agent chemotherapy was determined by the investigator before randomization from one of the following choices: eribulin (n=130), vinorelbine (n=63), gemcitabine (n=56), or capecitabine (n=22).1
Stratification1:
- Visceral metastasis (yes/no)
- Endocrine therapy in metastatic setting ≥6 months (yes/no)
- Prior lines of chemotherapy for metastatic disease (2 vs 3/4)
aDisease histology based on the ASCO/CAP criteria.3
bAdministration of TRODELVY was permitted beyond RECIST-defined disease progression if the patient was clinically stable and considered by the investigator to be deriving clinical benefit.1
The population of TROPiCS-02 has characteristics that may resemble those of your patients
DEMOGRAPHICS1 |
|
|---|---|
| Median age: | 56 years (range: 27−86 years); 26% ≥65 years |
| Sex: | 99% female |
| Race/ethnicity: | 67% White, 4% Black/African American, 3% Asian, 26% unknown |
DISEASE CHARACTERISTICS |
|
|---|---|
| ECOG performance status: | 0 (45%), 1 (55%)1 |
| Visceral metastases: | 95%1 |
| Median time from initial metastatic diagnosis: | 47.8 months2 |
| IHC status: | 52% HER2-low,c 40% HER2 IHC 0, 8% missing HER2 IHC status4 |
TREATMENT HISTORY1,2 |
|
|---|---|
| Prior systemic regimens: | Median of 7 (range: 3–17) prior systemic regimens in any setting and 3 (range 0–8) prior systemic chemotherapy regimens in the metastatic setting |
| Prior chemotherapy regimens: | ~42% received 2 prior chemotherapy regimens for metastatic disease; 58% received 3–4 prior chemotherapy regimens |
| Prior endocrine therapy: | 86% received prior endocrine therapy in the metastatic setting for ≥6 months |
| Prior CDK4/6i use: | 99% had prior CDK4/6i use in the metastatic setting, and 60% received it for ≤12 months |
c39 patients with HER2 IHC 2+ did not have ISH data documentation available for verification and were presumed to be HER2-low.4
CDK4/6i=cyclin-dependent kinase 4/6 inhibitor; ECOG=Eastern Cooperative Oncology Group; HER2=human epidermal growth factor receptor 2; IHC=immunohistochemistry; ISH=in situ hybridization.
TRODELVY demonstrated superior mPFS vs single-agent chemotherapy1
mPFS by BICR per RECIST 1.1 criteria (ITT population)1,a
VS
HR: 0.66 (95% CI: 0.53–0.83); P=0.0003
In a prespecified, descriptive analysis, the 12-month mPFS rate was 21% with TRODELVY (95% Cl: 15–28) vs 7% with single-agent chemotherapy (95% Cl: 3–14). Not powered for statistical analysis.2
aPFS is defined as the time from the date of randomization to the date of the first radiological disease progression or death due to any cause, whichever came first.1
TRODELVY gives you the power that may delay disease progression or death1,2
Consistent outcomes across key patient subgroups2
Limitation: These results are from a subgroup analysis of the Phase 3 TROPiCS-02 study. These endpoints were not powered for statistical analysis and should be considered descriptive only. Therefore, the results require cautious interpretation and could represent chance findings.2
mPFS by BICR based on RECIST 1.1 criteria and hazard ratio for disease progression or death2,e
Scroll left to review
Scroll left to review
ePFS is defined as the time from the date of randomization to the date of the first radiological disease progression or death due to any cause, whichever came first.1
fDefined as relapse to metastatic disease within 1 year of the end of (neo)adjuvant chemotherapy.2
Do your patients' characteristics align with these subgroups? TRODELVY may be the right next option for them.
TROPiCS-02 TRIAL VIDEO OVERVIEW
TRODELVY in pretreated HR+/HER2– mBC
With Dr. Martin Dietrich, MD, PhD, and Dr. Victoria Rizk, MD
TRODELVY: OL Video Series
HR+/HER2– in mBC Transcript
INDICATION
TRODELVY® (sacituzumab govitecan-hziy) is a Trop-2-directed antibody and topoisomerase inhibitor conjugate indicated for the treatment of adult patients with unresectable locally advanced or metastatic hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting.
IMPORTANT SAFETY INFORMATION
TRODELVY has a boxed warning.
TRODELVY can cause severe, life-threatening, or fatal neutropenia. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Primary prophylaxis with G-CSF is recommended for all patients at increased risk of febrile neutropenia. Initiate anti-infective treatment in patients with febrile neutropenia without delay.
TRODELVY can cause severe diarrhea. Monitor patients with diarrhea and give fluid and electrolytes as needed. At the onset of diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤Grade 1 and reduce subsequent doses.
Please see full Prescribing Information, including BOXED WARNING, at TRODELVYhcp.com.
VR: Hey, Dr. Dietrich. It’s great to see you again. I’m really looking forward to having a conversation today about the use of TRODELVY in pre-treated metastatic breast cancer patients, especially HR+/HER2–.
MD: Hello, Dr. Rizk. Thank you so much for making some time to meet with me today for this discussion.
VR: I don’t know about you, but it meant a great deal to me when the TROPiCS-02 data first came out. If it’s okay, why don’t we review the study design, and then we can talk about it after?
MD: That sounds great to me.
VR: The TROPiCS-02 trial was a Phase 3, randomized, active-controlled, open-label study that included a total of 543 patients with pretreated, unresectable, locally advanced or metastatic HR+/HER2– breast cancer. Patients were randomized one-to-one to receive either TRODELVY at 10 mg per kilogram IV on day one and day eight of a 21-day treatment cycle, or single-agent chemotherapy. 272 patients received TRODELVY, and 271 patients received single-agent chemotherapy. The choice of single-agent chemotherapy was determined by the investigator before randomization between eribulin, vinorelbine, gemcitabine, or capecitabine.
VR: For both groups, treatment was continued until disease progression or unacceptable toxicity. The primary endpoint in the trial was progression-free survival by blinded and independent central review based on the RECIST 1.1 criteria. Among the secondary endpoints, the ones we’ll look at today are overall survival and safety. So, after looking through the TROPiCS-02 trial for HR+/ HER2– metastatic breast cancer patients, what are your thoughts?
MD: I believe this was a well-needed trial in the later-line setting. And I think the trial design really reflected the patients that we are seeing, heavily pretreated, patients that have been on treatment for a long time. And I think that the choices of chemotherapy available were quite applicable to my practice. We’ve had capecitabine, gemcitabine, eribulin, vinorelbine, those are all choices that we would use in our clinic and are frequently challenged.
VR: I think it highlighted the need for an agent in this area because previously, we didn’t have a good understanding of which chemotherapy to even reach for and now we know chemotherapy may not be the right choice.
MD: So now that we reviewed the study design, maybe we can turn our attention to the efficacy results. Maybe, we can start with progression-free survival.
VR: Yeah, that’s a great idea.
MD: So the median progression-free survival in TROPiCS-02 was 5.5 months with TRODELVY versus 4.0 months with single-agent chemotherapy. In pretreated hormone receptor-positive, HER2-negative metastatic breast cancer across IHC status, TRODELVY provided a statistically significant and clinically meaningful PFS benefit. In fact, TRODELVY is the only Trop-2–directed ADC to show a statistically significant benefit in both median overall survival and progression-free survival. The median overall survival with TRODELVY was 14.4 months versus 11.2 months with single-agent chemotherapy. What do you think of these results?
VR: Well my biggest takeaway is that TRODELVY is the only Trop-2–directed antibody-drug conjugate with a statistically significant median progression-free survival and overall survival. It may not seem like a lot of time between 14 months and 11 months, but for patients, three months matters, and I think we owe them the ability to give them more time to the best of our ability. What do you think?
MD: Yeah, I agree. I think overall survival is the gold standard. I wasn’t positive that we would actually see positive overall survival benefit as it is so difficult to demonstrate in a later-line setting, but it really brought home the value of the drug.
VR: And it’s very reassuring to know that it was effective across HER2-negative IHC status because at least then we’re able to offer patients a therapy that regardless of what that number might be, because the biopsy might reflect only a certain area of the tumor and may not reflect the heterogeneity of the tumor profile. So being able to feel confident that you’re offering something that you know has good efficacy is very reassuring as an oncologist when you’re trying to give that confidence to your patients.
MD: For most of our patients, survival is really what we’re aiming for and extending the survival for as long as possible. And having seen a significant improvement in overall survival here that actually exceeded progression-free survival in numerical impact was actually quite valuable, so I think that’s where TRODELVY plays a significant role.
VR: It’s really interesting that you bring up a few points like overall survival. I think that’s the thing that patients really ask me, wanting to get a better understanding of how much time they have.
MD: Another concern is the hormone receptor-positive nature of the disease eventually becomes less relevant because we’ve exhausted endocrine therapy. Obviously, HER2 is part of the standard armamentarium for us to test for patients. We do see an evolution of HER2 as a biomarker. There are some variabilities in the readout. Not all pathologies provide the readout with the same level of granularity and certainty, so having the full spectrum of HER2 expression covered here in the non-amplified setting, I believe, is an important feature that gives us some reassurance about use in the HER2 non-amplified setting.
VR: So I think it might be worth going into the safety data just to talk through that. And then, we can talk through how you explain that to patients and what your expectations are for them.
MD: Oh, absolutely.
VR: Serious adverse reactions occurred in 28% of patients treated with TRODELVY. Here’s the list of serious reactions that occurred in more than 1% of patients.
VR: Shown here are the adverse reactions in 10% or more of patients with HR-positive, HER2-negative metastatic breast cancer in TROPiCS‑02. The most common adverse reactions, which were seen in 25% or more of patients including lab abnormalities, were decreased leukocytes, neutrophils, hemoglobin, and lymphocytes, diarrhea, fatigue, nausea, alopecia, increased glucose, constipation, and decreased albumin.
VR: So after reviewing those, what’s the way that you approach the conversation with patients about side effect management and what to expect?
MD: I think it’s an important discussion. In this context that we’re discussing, TROPiCS-02 patients already have been exposed to chemotherapy. I think some of the more common side effects and the individual experiences are familiar to them. We want to be as proactive as possible. I catch myself repeating myself over and over again to create a more lasting memory effect. And then obviously, to have an open channel of communication so that patients can reach out if there’s a question. How do you do it in your clinic?
VR: Yeah, very similar. So giving the patients just the knowledge that they need, I think makes the world of difference because they really just want an understanding of what should they plan for. And knowing when to reach out to their physician because we don’t want them to be at home, fearful of what’s to come, if they know what to expect, and if they plan ahead and they feel proactive. Metastatic breast cancer in general is something that makes people feel pretty powerless.
MD: I fully agree with that.
VR: Please keep watching for Important Safety Information.
IMPORTANT SAFETY INFORMATION
TRODELVY is contraindicated for patients with severe hypersensitivity reactions to TRODELVY.
Warnings and precautions for TRODELVY include neutropenia, diarrhea, hypersensitivity and infusion-related reactions, nausea and vomiting, increased risk of adverse reactions in patients with reduced UGT1A1 activity, and embryo-fetal toxicity.
Neutropenia: Severe, life-threatening, or fatal neutropenia can occur as early as the first cycle of treatment and may require dose modification. Neutropenia occurred in 64% of patients treated with TRODELVY. Grade 3-4 neutropenia occurred in 49% of patients. Febrile neutropenia occurred in 6%. Neutropenic colitis occurred in 1.4%. Primary prophylaxis with G-CSF is recommended starting in the first cycle of treatment in all patients at increased risk of febrile neutropenia, including older patients, patients with previous neutropenia, poor performance status, organ dysfunction, or multiple comorbidities. Monitor absolute neutrophil count (ANC) during treatment. Withhold TRODELVY for ANC below 1500/mm³ on Day 1 of any cycle or below 1000/mm³ on Day 8 of any cycle. Withhold TRODELVY for neutropenic fever. Treat neutropenia with G-CSF and administer prophylaxis in subsequent cycles as clinically indicated or indicated in Table 2 of USPI.
Diarrhea: Diarrhea occurred in 64% of all patients treated with TRODELVY. Grade 3-4 diarrhea occurred in 11% of patients. One patient had intestinal perforation following diarrhea. Diarrhea that led to dehydration and subsequent acute kidney injury occurred in 0.7% of all patients. Withhold TRODELVY for Grade 3-4 diarrhea and resume when resolved to ≤Grade 1. At onset, evaluate for infectious causes and if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (for example, fluid and electrolyte substitution) may also be employed as clinically indicated. Patients who exhibit an excessive cholinergic response to treatment can receive appropriate premedication (for example, atropine) for subsequent treatments.
Hypersensitivity and Infusion-Related Reactions: TRODELVY can cause serious hypersensitivity reactions including life-threatening anaphylactic reactions. Severe signs and symptoms included cardiac arrest, hypotension, wheezing, angioedema, swelling, pneumonitis, and skin reactions. Hypersensitivity reactions within 24 hours of dosing occurred in 35% of patients. Grade 3-4 hypersensitivity occurred in 2% of patients. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.2%. The incidence of anaphylactic reactions was 0.2%. Pre-infusion medication is recommended. Have medications and emergency equipment to treat such reactions available for immediate use. Observe patients closely for hypersensitivity and infusion-related reactions during each infusion and for at least 30 minutes after completion of each infusion. Permanently discontinue TRODELVY for Grade 4 infusion-related reactions.
Nausea and Vomiting: TRODELVY is emetogenic and can cause severe nausea and vomiting. Nausea occurred in 64% of all patients treated with TRODELVY and Grade 3-4 nausea occurred in 3% of these patients. Vomiting occurred in 35% of patients and Grade 3-4 vomiting occurred in 2% of these patients. Premedicate with a two or three drug combination regimen (for example, dexamethasone with either a 5-HT3 receptor antagonist or an NK1 receptor antagonist as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting (CINV). Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting and resume with additional supportive measures when resolved to ≤Grade 1. Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity: Patients homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia and may be at increased risk for other adverse reactions with TRODELVY. The incidence of Grade 3-4 neutropenia was 58% in patients homozygous for the UGT1A1*28, 49% in patients heterozygous for the UGT1A1*28 allele, and 43% in patients homozygous for the wild-type allele. The incidence of Grade 3-4 anemia was 21% in patients homozygous for the UGT1A1*28 allele, 10% in patients heterozygous for the UGT1A1*28 allele, and 9% in patients homozygous for the wild-type allele. Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on clinical assessment of the onset, duration and severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 function.
Embryo-Fetal Toxicity: Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
The following adverse reactions were observed with TRODELVY.
In the pooled safety population, the most common (≥ 25%) adverse reactions including laboratory abnormalities were decreased leukocyte count (84%), decreased neutrophil count (75%), decreased hemoglobin (69%), diarrhea (64%), nausea (64%), decreased lymphocyte count (63%), fatigue (51%), alopecia (45%), constipation (37%), increased glucose (37%), decreased albumin (35%), vomiting (35%), decreased appetite (30%), decreased creatinine clearance (28%), increased alkaline phosphatase (28%), decreased magnesium (27%), decreased potassium (26%), and decreased sodium (26%).
In the TROPiCS-02 study (locally advanced or metastatic HR-positive, HER2-negative breast cancer), the most common adverse reactions (incidence ≥25%) were diarrhea, fatigue, nausea, alopecia, and constipation. The most frequent serious adverse reactions (incidence >1%) were diarrhea (5%), febrile neutropenia (4%), neutropenia (3%), abdominal pain, colitis, neutropenic colitis, pneumonia, and vomiting (each 2%). Serious adverse reactions were reported in 28% of patients, and 6% discontinued therapy due to adverse reactions. The most common Grade 3-4 lab abnormalities (incidence ≥25%) in the TROPiCS-02 study were reduced neutrophils and leukocytes.
Drug interactions for TRODELVY include UGT1A1 inhibitors and UGT1A1 inducers.
UGT1A1 Inhibitors: Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38. Avoid administering UGT1A1 inhibitors with TRODELVY.
UGT1A1 Inducers: Exposure to SN-38 may be reduced in patients concomitantly receiving UGT1A1 enzyme inducers. Avoid administering UGT1A1 inducers with TRODELVY.
Please see full Prescribing Information, including BOXED WARNING, at TRODELVYhcp.com.
This information does not constitute the provision of medical advice and should not substitute for clinical decision-making.
Presenters are paid consultants of Gilead.
TRODELVY, the TRODELVY logo, GILEAD, and the GILEAD logo are trademarks of Gilead Sciences, Inc., or its related companies. All other marks are the property of their respective owners.
© 2026 Gilead Sciences, Inc. All rights reserved. US-TROP-2131 01/26
ASCO=American Society of Clinical Oncology; BICR=blinded independent central review; CAP=College of American Pathologists; CDK=cyclin-dependent kinase; CI=confidence interval; DOR=duration of response; HER2-=human epidermal growth factor receptor 2–negative; HR=hazard ratio; HR+=hormone receptor–positive; IHC=immunohistochemistry; ISH=in situ hybridization; ITT=intent-to-treat; mBC=metastatic breast cancer; mPFS=median progression-free survival; NE=not evaluable; ORR=overall response rate; OS=overall survival; PFS=progression-free survival; PRO=patient-reported outcome; RECIST=Response Evaluation Criteria in Solid Tumors.
References: 1. TRODELVY. Prescribing Information. Gilead Sciences, Inc.; 2026. 2. Rugo HS, Bardia A, Marmé F, et al. Sacituzumab govitecan in hormone receptor–positive/human epidermal growth factor receptor 2–negative metastatic breast cancer. J Clin Oncol. 2022;40(29):3365-3376. 3. Immunomedics, Inc. Phase 3 study of sacituzumab govitecan (IMMU-132) versus treatment of physician’s choice (TPC) in subjects with hormonal receptor-positive (HR+) human epidermal growth factor receptor 2 (HER2) negative metastatic breast cancer (MBC) who have failed at least two prior chemotherapy regimens. Published December 21, 2018. Accessed January 5, 2024. https://ascopubs.org/doi/suppl/10.1200/JCO.22.01002/suppl_file/protocol_JCO.22.01002.pdf 4. Schmid P, Cortes J, Marmé F, et al. Sacituzumab govitecan efficacy in HR+/HER2- metastatic breast cancer by HER2 immunohistochemistry status in the phase 3 TROPiCS-02 study. Presented at: European Society for Medical Oncology (ESMO) Congress; September 9-13, 2022; Paris, France. Presentation FPN 214MO.
Continue exploring HR+/HER2– mBC efficacy
Select another topic
Scroll left to select an efficacy measure
TRODELVY® (sacituzumab govitecan-hziy) is a Trop-2–directed antibody and topoisomerase inhibitor conjugate indicated in adult patients:
Locally Advanced or Metastatic Triple-Negative Breast Cancer
First Line
- As a single agent for the first-line treatment of unresectable locally advanced or metastatic triple-negative breast cancer (mTNBC) who are not candidates for PD-1 or PD-L1 inhibitor-based therapy
- In combination with pembrolizumab or pembrolizumab and berahyaluronidase alfa-pmph for the first-line treatment of unresectable locally advanced or mTNBC whose tumors express PD-L1 [Combined Positive Score (CPS ≥10)] as determined by an FDA-authorized test
Second Line or Later
- For the treatment of unresectable locally advanced or mTNBC who have received two or more prior systemic therapies, at least one of them for metastatic disease.
Locally Advanced or Metastatic HR-positive, HER2-negative Breast Cancer
- For the treatment of unresectable locally advanced or metastatic hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+, or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting.
Important Safety Information
Tap for Important Safety Information, including BOXED WARNING: Neutropenia and Diarrhea.
Boxed Warning: neutropenia and diarrhea
- TRODELVY can cause severe, life-threatening, or fatal neutropenia. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Primary prophylaxis with G-CSF is recommended for all patients at increased risk of febrile neutropenia. Initiate anti-infective treatment in patients with febrile neutropenia without delay.
- TRODELVY can cause severe diarrhea. Monitor patients with diarrhea and give fluid and electrolytes as needed. At the onset of diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤Grade 1 and reduce subsequent doses.
Contraindications
- Severe hypersensitivity reaction to TRODELVY.
Warnings and precautions
Neutropenia: Severe, life-threatening, or fatal neutropenia can occur as early as the first cycle of treatment and may require dose modification. Neutropenia occurred in 64% of patients treated with TRODELVY. Grade 3-4 neutropenia occurred in 48% of patients. Febrile neutropenia occurred in 6%. Neutropenic colitis occurred in 1.4%. Primary prophylaxis with G-CSF is recommended starting in the first cycle of treatment in all patients at increased risk of febrile neutropenia, including older patients, patients with previous neutropenia, poor performance status, organ dysfunction, or multiple comorbidities. Monitor absolute neutrophil count (ANC) during treatment. Withhold TRODELVY for ANC below 1500/mm3 on Day 1 of any cycle or below 1000/mm3 on Day 8 of any cycle. Withhold TRODELVY for neutropenic fever. Treat neutropenia with G-CSF and administer prophylaxis in subsequent cycles as clinically indicated or indicated in Table 2 of USPI.
Diarrhea: Diarrhea occurred in 62% of all patients treated with TRODELVY. Grade 3-4 diarrhea occurred in 10% of patients. One patient had intestinal perforation following diarrhea. Diarrhea that led to dehydration and subsequent acute kidney injury occurred in 0.6% of all patients. Withhold TRODELVY for Grade 3-4 diarrhea and resume when resolved to ≤Grade 1. At onset, evaluate for infectious causes and, if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (eg, fluid and electrolyte replacement) may also be employed as clinically indicated. Patients who exhibit an excessive cholinergic response to treatment can receive appropriate premedication (eg, atropine) for subsequent treatments.
Hypersensitivity and Infusion-Related Reactions: TRODELVY can cause serious hypersensitivity reactions, including life-threatening anaphylactic reactions. Severe signs and symptoms included cardiac arrest, hypotension, wheezing, angioedema, swelling, and skin reactions. Hypersensitivity reactions occurred in 28% of patients with 13% occurring within 24 hours of dosage. Grade 3-4 hypersensitivity occurred in 1.5% of patients with 0.4% of these occurring within 24 hours of dosage. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.4%. The incidence of anaphylactic reaction was <0.1%. Pre-infusion medication is recommended. Have medications and emergency equipment to treat such reactions available for immediate use. Closely monitor patients for hypersensitivity and infusion-related reactions during each infusion and for at least 30 minutes after completion of each infusion. Permanently discontinue TRODELVY for Grade 4 infusion-related reactions.
Nausea and Vomiting: TRODELVY is emetogenic and can cause severe nausea and vomiting. Nausea occurred in 63% of all patients treated with TRODELVY, and Grade 3-4 nausea occurred in 3% of these patients. Vomiting occurred in 33% of patients, and Grade 3-4 vomiting occurred in 2% of these patients. Premedicate with a two- or three-drug combination regimen (eg, dexamethasone with either a 5-HT3 receptor antagonist or an NK1 receptor antagonist, as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting. Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting and resume with additional supportive measures when resolved to ≤Grade 1. Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity: Patients homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia and may be at increased risk for other adverse reactions with TRODELVY. The incidence of Grade 3-4 neutropenia was 57% in patients homozygous for the UGT1A1*28 allele, 48% in patients heterozygous for the UGT1A1*28 allele, and 41% in patients homozygous for the wild-type allele. The incidence of Grade 3-4 anemia was 17% in patients homozygous for the UGT1A1*28 allele, 9% in patients heterozygous for the UGT1A1*28 allele, and 8% in patients homozygous for the wild-type allele. Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on clinical assessment of the onset, duration, and severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 function.
Embryo-Fetal Toxicity: Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
Adverse Reactions
In the pooled safety population of TRODELVY as a single agent, the most common (≥25%) adverse reactions, including laboratory abnormalities, were decreased leukocyte count (83%), decreased neutrophil count (77%), decreased hemoglobin (71%), nausea (63%), diarrhea (62%), decreased lymphocyte count (60%), fatigue (59%), alopecia (47%), increased glucose (40%), constipation (37%), vomiting (33%), decreased albumin (32%), increased alkaline phosphatase (30%), decreased appetite (28%), abdominal pain (27%), decreased creatinine clearance (27%), decreased magnesium and potassium (26% each).
In the safety population of TRODELVY in combination with pembrolizumab, the most common (≥25%) adverse reactions, including laboratory abnormalities, were decreased neutrophil count and hemoglobin (86% each), decreased leukocyte count (84%), diarrhea (72%), nausea (68%), decreased lymphocyte count (61%), fatigue (58%), alopecia (52%), increased alkaline phosphatase and glucose (50% each), increased alanine aminotransferase (47%), constipation (41%), increased aspartate aminotransferase (40%), rash (37%), decreased potassium (35%), increased lactate dehydrogenase (34%), vomiting (29%), abdominal pain, headache, and increased eosinophils (26% each), and decreased albumin (25%).
In the ASCENT-03 study (single agent in previously untreated, unresectable locally advanced or mTNBC), the most common adverse reactions (incidence ≥25%) were nausea, diarrhea, alopecia, fatigue, constipation, and vomiting. The most frequent serious adverse reactions (SAR) (>2%) were diarrhea, febrile neutropenia, and neutropenia (3.6% each), and pneumonia (2.9%). SAR occurred in 26% of patients, and 3.6% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 2.5% of patients and included sepsis (1.1%), and acute respiratory failure, neutropenic colitis, pneumonia, and septic shock (0.4% each). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils and leukocytes.
In the ASCENT-04 study (in combination with pembrolizumab in previously untreated, unresectable locally advanced or mTNBC whose tumors express PD-L1), the most common adverse reactions (incidence ≥25%) were diarrhea, nausea, fatigue, alopecia, constipation, rash, vomiting, abdominal pain, and headache. The most frequent SAR (≥2%) were febrile neutropenia (7%), neutropenia (6%), diarrhea (5%), and fatigue and pneumonia (2.3% each). SAR occurred in 38% of patients, and 7% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 3.2% of patients and included death (unknown cause) (0.9%) and completed suicide, neutropenic sepsis, sepsis, pneumonia, and pulmonary embolism (0.5% each). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils and leukocytes.
In the ASCENT study (previously treated locally advanced or mTNBC), the most common adverse reactions (incidence ≥25%) were fatigue, diarrhea, nausea, alopecia, constipation, vomiting, abdominal pain, and decreased appetite. The most frequent SAR (>1%) were neutropenia (7%), diarrhea (4%), and pneumonia (3%). SAR occurred in 27% of patients, and 5% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 1.2% of patients and included respiratory failure (0.8%) and pneumonia (0.4%). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils, leukocytes, and lymphocytes.
In the TROPiCS-02 study (locally advanced or metastatic HR+/HER2– breast cancer), the most common adverse reactions (incidence ≥25%) were diarrhea, fatigue, nausea, alopecia, and constipation. The most frequent SAR (>1%) were diarrhea (5%), febrile neutropenia (4.1%), neutropenia (3%), abdominal pain (2.2%), neutropenic colitis and vomiting (1.9% each), and colitis and pneumonia (1.5% each). SAR occurred in 28% of patients, and 6% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 2.2% of patients and included arrhythmia, COVID-19 pneumonia, pneumonia, nervous system disorder, pulmonary embolism, and septic shock (0.4% each). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils and leukocytes.
Drug Interactions
UGT1A1 Inhibitors: Avoid administering UGT1A1 inhibitors with TRODELVY. SN-38 is a UGT1A1 substrate. Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38.
UGT1A1 Inducers: Avoid administering UGT1A1 inducers with TRODELVY. SN-38 is a UGT1A1 substrate. Concomitant administration of TRODELVY with inducers of UGT1A1 may reduce exposure to SN-38.
Please see full Prescribing Information, including BOXED WARNING.

TRODELVY® (sacituzumab govitecan-hziy) is a Trop-2–directed antibody and topoisomerase inhibitor conjugate indicated in adult patients:
Locally Advanced or Metastatic Triple-Negative Breast Cancer
First Line
- As a single agent for the first-line treatment of unresectable locally advanced or metastatic triple-negative breast cancer (mTNBC) who are not candidates for PD-1 or PD-L1 inhibitor-based therapy
- In combination with pembrolizumab or pembrolizumab and berahyaluronidase alfa-pmph for the first-line treatment of unresectable locally advanced or mTNBC whose tumors express PD-L1 [Combined Positive Score (CPS ≥10)] as determined by an FDA-authorized test
Second Line or Later
- For the treatment of unresectable locally advanced or mTNBC who have received two or more prior systemic therapies, at least one of them for metastatic disease.
Locally Advanced or Metastatic HR-positive, HER2-negative Breast Cancer
- For the treatment of unresectable locally advanced or metastatic hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+, or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting.
Important Safety Information
Tap for Important Safety Information, including BOXED WARNING: Neutropenia and Diarrhea.
Boxed Warning: neutropenia and diarrhea
- TRODELVY can cause severe, life-threatening, or fatal neutropenia. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Primary prophylaxis with G-CSF is recommended for all patients at increased risk of febrile neutropenia. Initiate anti-infective treatment in patients with febrile neutropenia without delay.
- TRODELVY can cause severe diarrhea. Monitor patients with diarrhea and give fluid and electrolytes as needed. At the onset of diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤Grade 1 and reduce subsequent doses.
Contraindications
- Severe hypersensitivity reaction to TRODELVY.
Warnings and precautions
Neutropenia: Severe, life-threatening, or fatal neutropenia can occur as early as the first cycle of treatment and may require dose modification. Neutropenia occurred in 64% of patients treated with TRODELVY. Grade 3-4 neutropenia occurred in 48% of patients. Febrile neutropenia occurred in 6%. Neutropenic colitis occurred in 1.4%. Primary prophylaxis with G-CSF is recommended starting in the first cycle of treatment in all patients at increased risk of febrile neutropenia, including older patients, patients with previous neutropenia, poor performance status, organ dysfunction, or multiple comorbidities. Monitor absolute neutrophil count (ANC) during treatment. Withhold TRODELVY for ANC below 1500/mm3 on Day 1 of any cycle or below 1000/mm3 on Day 8 of any cycle. Withhold TRODELVY for neutropenic fever. Treat neutropenia with G-CSF and administer prophylaxis in subsequent cycles as clinically indicated or indicated in Table 2 of USPI.
Diarrhea: Diarrhea occurred in 62% of all patients treated with TRODELVY. Grade 3-4 diarrhea occurred in 10% of patients. One patient had intestinal perforation following diarrhea. Diarrhea that led to dehydration and subsequent acute kidney injury occurred in 0.6% of all patients. Withhold TRODELVY for Grade 3-4 diarrhea and resume when resolved to ≤Grade 1. At onset, evaluate for infectious causes and, if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (eg, fluid and electrolyte replacement) may also be employed as clinically indicated. Patients who exhibit an excessive cholinergic response to treatment can receive appropriate premedication (eg, atropine) for subsequent treatments.
Hypersensitivity and Infusion-Related Reactions: TRODELVY can cause serious hypersensitivity reactions, including life-threatening anaphylactic reactions. Severe signs and symptoms included cardiac arrest, hypotension, wheezing, angioedema, swelling, and skin reactions. Hypersensitivity reactions occurred in 28% of patients with 13% occurring within 24 hours of dosage. Grade 3-4 hypersensitivity occurred in 1.5% of patients with 0.4% of these occurring within 24 hours of dosage. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.4%. The incidence of anaphylactic reaction was <0.1%. Pre-infusion medication is recommended. Have medications and emergency equipment to treat such reactions available for immediate use. Closely monitor patients for hypersensitivity and infusion-related reactions during each infusion and for at least 30 minutes after completion of each infusion. Permanently discontinue TRODELVY for Grade 4 infusion-related reactions.
Nausea and Vomiting: TRODELVY is emetogenic and can cause severe nausea and vomiting. Nausea occurred in 63% of all patients treated with TRODELVY, and Grade 3-4 nausea occurred in 3% of these patients. Vomiting occurred in 33% of patients, and Grade 3-4 vomiting occurred in 2% of these patients. Premedicate with a two- or three-drug combination regimen (eg, dexamethasone with either a 5-HT3 receptor antagonist or an NK1 receptor antagonist, as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting. Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting and resume with additional supportive measures when resolved to ≤Grade 1. Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity: Patients homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia and may be at increased risk for other adverse reactions with TRODELVY. The incidence of Grade 3-4 neutropenia was 57% in patients homozygous for the UGT1A1*28 allele, 48% in patients heterozygous for the UGT1A1*28 allele, and 41% in patients homozygous for the wild-type allele. The incidence of Grade 3-4 anemia was 17% in patients homozygous for the UGT1A1*28 allele, 9% in patients heterozygous for the UGT1A1*28 allele, and 8% in patients homozygous for the wild-type allele. Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on clinical assessment of the onset, duration, and severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 function.
Embryo-Fetal Toxicity: Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
Adverse Reactions
In the pooled safety population of TRODELVY as a single agent, the most common (≥25%) adverse reactions, including laboratory abnormalities, were decreased leukocyte count (83%), decreased neutrophil count (77%), decreased hemoglobin (71%), nausea (63%), diarrhea (62%), decreased lymphocyte count (60%), fatigue (59%), alopecia (47%), increased glucose (40%), constipation (37%), vomiting (33%), decreased albumin (32%), increased alkaline phosphatase (30%), decreased appetite (28%), abdominal pain (27%), decreased creatinine clearance (27%), decreased magnesium and potassium (26% each).
In the safety population of TRODELVY in combination with pembrolizumab, the most common (≥25%) adverse reactions, including laboratory abnormalities, were decreased neutrophil count and hemoglobin (86% each), decreased leukocyte count (84%), diarrhea (72%), nausea (68%), decreased lymphocyte count (61%), fatigue (58%), alopecia (52%), increased alkaline phosphatase and glucose (50% each), increased alanine aminotransferase (47%), constipation (41%), increased aspartate aminotransferase (40%), rash (37%), decreased potassium (35%), increased lactate dehydrogenase (34%), vomiting (29%), abdominal pain, headache, and increased eosinophils (26% each), and decreased albumin (25%).
In the ASCENT-03 study (single agent in previously untreated, unresectable locally advanced or mTNBC), the most common adverse reactions (incidence ≥25%) were nausea, diarrhea, alopecia, fatigue, constipation, and vomiting. The most frequent serious adverse reactions (SAR) (>2%) were diarrhea, febrile neutropenia, and neutropenia (3.6% each), and pneumonia (2.9%). SAR occurred in 26% of patients, and 3.6% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 2.5% of patients and included sepsis (1.1%), and acute respiratory failure, neutropenic colitis, pneumonia, and septic shock (0.4% each). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils and leukocytes.
In the ASCENT-04 study (in combination with pembrolizumab in previously untreated, unresectable locally advanced or mTNBC whose tumors express PD-L1), the most common adverse reactions (incidence ≥25%) were diarrhea, nausea, fatigue, alopecia, constipation, rash, vomiting, abdominal pain, and headache. The most frequent SAR (≥2%) were febrile neutropenia (7%), neutropenia (6%), diarrhea (5%), and fatigue and pneumonia (2.3% each). SAR occurred in 38% of patients, and 7% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 3.2% of patients and included death (unknown cause) (0.9%) and completed suicide, neutropenic sepsis, sepsis, pneumonia, and pulmonary embolism (0.5% each). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils and leukocytes.
In the ASCENT study (previously treated locally advanced or mTNBC), the most common adverse reactions (incidence ≥25%) were fatigue, diarrhea, nausea, alopecia, constipation, vomiting, abdominal pain, and decreased appetite. The most frequent SAR (>1%) were neutropenia (7%), diarrhea (4%), and pneumonia (3%). SAR occurred in 27% of patients, and 5% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 1.2% of patients and included respiratory failure (0.8%) and pneumonia (0.4%). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils, leukocytes, and lymphocytes.
In the TROPiCS-02 study (locally advanced or metastatic HR+/HER2– breast cancer), the most common adverse reactions (incidence ≥25%) were diarrhea, fatigue, nausea, alopecia, and constipation. The most frequent SAR (>1%) were diarrhea (5%), febrile neutropenia (4.1%), neutropenia (3%), abdominal pain (2.2%), neutropenic colitis and vomiting (1.9% each), and colitis and pneumonia (1.5% each). SAR occurred in 28% of patients, and 6% permanently discontinued TRODELVY due to adverse reactions. Fatal adverse reactions occurred in 2.2% of patients and included arrhythmia, COVID-19 pneumonia, pneumonia, nervous system disorder, pulmonary embolism, and septic shock (0.4% each). The most common Grade 3-4 lab abnormalities (incidence ≥25%) were decreased neutrophils and leukocytes.
Drug Interactions
UGT1A1 Inhibitors: Avoid administering UGT1A1 inhibitors with TRODELVY. SN-38 is a UGT1A1 substrate. Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38.
UGT1A1 Inducers: Avoid administering UGT1A1 inducers with TRODELVY. SN-38 is a UGT1A1 substrate. Concomitant administration of TRODELVY with inducers of UGT1A1 may reduce exposure to SN-38.
Please see full Prescribing Information, including BOXED WARNING.